
9,10-菲醌选择性溴化反应研究
2016-07-14 02:00杨汉民沈千艳刘长玉
中南民族大学学报(自然科学版) 2016年2期
杨汉民,沈千艳,刘长玉,郑 文,陶 俊
(1中南民族大学 化学与材料科学学院,武汉 430074;2 武汉朋和科技有限公司,武汉 430074)
9,10-菲醌选择性溴化反应研究
杨汉民1,沈千艳1,刘长玉1,郑文2,陶俊2
(1中南民族大学 化学与材料科学学院,武汉 430074;2 武汉朋和科技有限公司,武汉 430074)
摘要通过改变反应物的投料比、溶剂、反应温度等因素高选择性合成了9,10-菲醌系列溴化物. 结果表明:9,10-菲醌与N-溴代丁酰亚胺摩尔比为1.0∶1.4,在浓硫酸中0 ℃反应3 h得到了2-溴-9,10-菲醌,收率60%;在相同条件下,9,10-菲醌与N-溴代丁酰亚胺摩尔比为1.0∶2.3得到了2,7-二溴-9,10-菲醌,收率76%;9,10-菲醌与溴素摩尔比为1.0∶1.4,过氧化苯甲酰为引发剂,在冰乙酸中106 ℃反应3 h,得到了3-溴-9,10-菲醌,收率85%,在相同条件下,9,10-菲醌与溴素摩尔比为1.0∶3.4,得到了3,6-二溴-9,10-菲醌,收率90%. 对9,10-菲醌的选择性溴化规律进行了探讨,发现溴化剂的加入量是决定生成单溴代或双溴代产物的主要因素.
关键词9,10-菲醌;菲醌溴代衍生物;溴化反应;溴化剂;选择性
溴代菲醌衍生物是9,10-菲醌的一种重要菲醌类衍生物,由于独特的性质使其在光化学、分析化学和生物有机化学等领域备受关注[1,2].溴代菲醌衍生物作为合成有机发光二极管(OLED)材料的一类重要中间体在菲醌衍生物中也脱颖而出,成为备受关注的焦点.OLED材料具有自发光的特性,具有许多发光二极管(LED)材料不可比拟的优势,有较大发展潜力和市场前景,溴代菲醌类衍生物的合成及性质研究也成为了研究热点之一.
对溴代菲醌衍生物的合成研究较多,如Song, Yabin等[5-8]以9,10-菲醌为原料,在PhNO2作溶剂的条件下与溴素反应得到了产率74.9%的3-溴-9,10-菲醌,产率85.1%的3,6-二溴-9,10-菲醌.Fomina, Nadezda等[10]以PhNO2作溶剂,9,10-菲醌为原料,过氧化苯甲酰为引发剂加入溴素得到了3位的单溴代的菲醌衍生物.以上实验虽产率较高,却采用高温下易燃易爆有毒的硝基苯作溶剂,工业化生产有一定的困难. Schmidhalter, Beat等[9]在Fomina, Nadezda基础上改进,摒弃易燃易爆有毒的溶剂PhNO2选取乙酸作溶剂106 ℃反应3 h,室温反应20 h,产品用热的N,N-二甲基甲酰胺重结晶,热的甲醇和正己烷洗后,合成了产率61%的3-溴-9,10-菲醌. Fomina, Nadezda等合成缺点是室温反应过长,产率不高;Muddasir Hanif等[3]对合成双溴代菲醌衍生物进行了报道,选用9,10-菲二酮为原料,浓硫酸作溶剂于室温下,加入N-溴代丁酰亚胺制备2,7-二溴-9,10-菲醌,获得52%的收率.
本文重复了Muddasir Hanif等[3]的实验后,发现合成方法简单并通过控制N-溴代丁酰亚胺的加入量,将收率提高到了76.8%.对于合成2-溴-9,10-菲醌和3,6-二溴-9,10-菲醌文献报道较少,由于溴代菲醌衍生物的对称性强,溶解性差,熔沸点高,制备十分困难,多数合成方法及后处理较复杂繁琐.基于简单经济环保的原则,本文拟通过一系列的实验,探索9,10-菲醌的选择性溴化规律,并对其合成工艺进行优化,为工业化生产提供数据支持.其具体合成路线如图1.
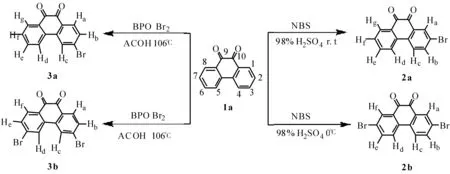
图1 溴代菲醌衍生物合成路线Fig.1 Synthetic routes of bromine phenanthrenequinone derivatives
1实验部分
1.1试剂和仪器
9,10-菲醌、溴素、N-溴代丁酰亚胺(NBS)、98% H2SO4、冰乙酸(武汉格奥化学有限公司),过氧化苯甲酰(BPO)、无水乙醇、二氯甲烷(DCM)、N,N-二甲基甲酰胺(DMF)、无水甲醇、无水乙醚(国药集团化学试剂有限公司),以上试剂均为AR.
核磁共振仪(Bruker AV 400 MHz型, CDCl3或DMSO作溶剂, TMS作内标),暗箱式三用紫外分析仪(ZF-7型,上海嘉鹏科技有限公司),高功率数控超声波清洗器(KQ-400KD型, 济宁天华超声电子仪器有限公司),数显恒温磁力搅拌油浴锅(DF-2型, 河南巩义予华仪器有限责任公司),台式真空干燥箱(DZF-6050, 河南巩义予华仪器有限责任公司).
1.22-溴-9,10-菲醌(2a)的合成
在带有磁力搅拌器的250 mL三口瓶里加入9,10-菲醌5.0 g(24.0 mmol), 98%浓硫酸150 mL,搅拌5 min,0 ℃下加入6.84 g(38.4 mmol) NBS(分3次加入, 每隔45 min加入2.28 g),冰水浴下反应3 h后,将混合液倒入100 mL冰水中,搅拌30 min,过滤,滤饼用100 mL乙醇加热回流3 h后热过滤,粗产品再用250 mL二氯甲烷加热回流热过滤,得到4.2 g橙红色固体2-溴-9,10-菲醌(2a),收率60%.1H NMR(CDCl3, 400 MHz)δ:8.30(d,J=2.4 Hz, 1H, -Ha), 8.21(dd,J1= 7.6 Hz,J2= 1.6 Hz ,1 H, -Hb ), 7.98(d,J=8 Hz, 1 H, -Hc), 7.90(d,J=8.6 Hz, 1H, -Hg), 7.83(dd,J1= 8.4 Hz,J2= 2.0 Hz, 1 H, -Hd), 7.74(t,J=16.8 Hz, 1 H, -Hf), 7.51(t,J=16 Hz, 1H, -He).
1.32,7-二溴-9,10-菲醌(2b)的合成
在带有磁力搅拌的三口烧瓶中,加入9,10-菲醌5.0 g (24.0 mmol),98%浓硫酸180 mL,搅拌5 min,加入NBS 10 g(56.1 mmol),室温下搅拌3 h,将反应混合物迅速倒入100 mL冰水中搅拌30 min,有橘红色固体析出,过滤,滤饼用冰水洗涤,粗产品用DMSO重结晶后,得到6.7 g 橘红色固体 2,7-二溴-9,10-菲醌(2b), 收率76.8%.1H NMR(d6-DMSO, 400 MHz)δ:8.24(dd,J1= 8.8 Hz,J2=0.4 Hz, 2 H, -Hb, -He) , 8.06(d,J=2 Hz, 2 H, -Ha ,-Hf), 7.95(dd,J1= 8 Hz,J2= 2.0 Hz, 2 H, - Hc, -Hd)与文献[3]相符.
1.43-溴-9,10-菲醌(3a)的合成
向带有磁力搅拌的干燥三口烧瓶中加入9,10-菲二酮20.8 g(0.1 mol)、乙酸250 mL、BPO 1.2 g(0.005 mol),油浴加热到106 ℃使其溶解,磁力搅拌,分批滴加溴素24 g(0.15 mol),滴完后反应3 h,趁热过滤,得粗产品,用乙醇洗涤粗产品,用DMF重结晶,在DCM回流5 h后过滤,固体经真空干燥,得4.9 g黄色粉末3-溴-9,10-菲醌(3a),收率85%.1H NMR(CDCl3, 400 MHz)δ: 8.22(dd,J1= 8 Hz,J2= 1.6 Hz, 1H , -Hb), 8.175(d,J= 2 Hz, 1H, -Hc), 8.05(d,J=8 Hz, 1H , -Ha), 7.98(d,J=8 Hz, 1H , -Hg), 7.75(t,J=16 Hz, 1H, -Hf), 7.62(dd,J1= 4 Hz,J2= 1.6 Hz, 1H, -He), 7.525(t,J=12 Hz, 1H, -Hd)与文献[7]相符.
1.53,6-二溴-9,10-菲醌(3b)的合成
干燥条件下,将9,10-菲醌5.0 g(24 mmol),冰乙酸60 mL和BPO 0.29 g(1.2 mmol)加入到100 mL带有磁力搅拌的三口瓶中,加热106 ℃使原料溶解后,一次性加入溴素11.5 g(72.1 mmol),待产物生成后再反应3 h,停止加热,热过滤,粗产品先用乙醇洗2~3次,加入30 mL无水乙醇回流5 h以上,可滴加少量乙酸,热过滤,滤饼用30 mL DCM回流5 h,热过滤;得到6.8 g黄色固体3,6-二溴-9,10-菲醌(3b),收率90%.1H NMR(CDCl3, 400 MHz)δ:8.12(d,J=1.6 Hz, 2 H, -Hc ,-Hd ), 8.075(d,J=12 Hz, 2 H, -Ha,-Hf), 7.67(dd,J1= 8.4 Hz,J2= 1.6 Hz, 2 H, -Hb, -He).
2结果与讨论
此类化合物的合成要在酸性条件下进行,酸的存在能提高自由基引发的速率,促使反应向正反应方向进行,实验中发现合成2a和2b宜用浓硫酸而合成3a和3b宜用冰乙酸,同时也通过多组实验找到了最佳反应时间为3 h;2a和2b的反应温度分别为0℃和室温;3a和3b为106 ℃,但是不论是合成2a还是3a都应该控制好9,10-菲醌与NBS或溴素的摩尔比(一般1.0∶1.3 ~ 1.0∶1.6 为宜),且NBS和溴素应分批缓慢加入,以避免大量副产物2b和3b的生成导致后续纯化困难.
2.12-溴-9,10-菲醌(2a)和2,7-二溴-9,10-菲醌(2b)的控制合成
2a和2b的合成中以浓硫酸作溶剂,以NBS作溴代物,加入不同摩尔的NBS分别得到了2a和2b,尽管NBS溴代反应是以分子溴为反应实体,但仍以自由基取代为主[10].
合成2a时,0 ℃下每隔45 min分3次加入NBS;而合成2b则需在室温下,直接一次性加入所需的NBS,反应时间均为3 h.在最佳条件下,不同的9,10-菲醌与NBS的摩尔比值对反应产率的影响不同,分别选用了1.0∶0.9 ~ 1.0∶2.8的10组实验数据进行讨论,其中1.0∶0.9 ~ 1.0∶1.8时的5组实验的温度为0 ℃,1.0∶2.0 ~ 1.0∶2.8时5组实验的温度为室温,结果如图3. 图3结果表明:在温度和他条件不变时,9,10-菲醌与NBS的摩尔比为1.0∶1.4 时,2a的收率高达60%,简便了后处理过程;9,10-菲醌与NBS的摩尔比为1.0∶2.3时,2b的收率高达70%,再增加NBS当量对目标产品产率无太大提高,故合成3a最佳摩尔比为1.0∶1.4,NBS需分批加入.合成2b时最佳摩尔比为1.0∶2.3,NBS需一次性加入.另外,延长反应时间并未发现产率有所提高.

图2 NBS与9,10-菲醌的物质的量比值与收率的关系Fig.2 The relationship between the molar ratio ofN-bromobutanimide to 9,10-phenanthrenequinoneand their yields
2.23-溴-9,10-菲醌(3a)和3,6-二溴-9,10-菲醌(3b)的控制合成
3a和3b是在冰乙酸中加入自由基引发剂BPO和溴素反应3 h后获得,其推测其反应历程如下.

通过多组单因素实验,找到了最佳反应温度为106 ℃,反应时间为3 h.在最佳温度和时间的条件下,找到9,10-菲醌与溴素的摩尔比与反应收率之间的关系,选择9,10-菲醌与溴素的摩尔比从1.0∶1.0 ~ 1.0∶3.4的10组实验数据进行了讨论,结果如图3. 图3结果表明:在温度及其他条件不变情况下,菲醌与溴素最佳摩尔比为1.0∶1.4,反应时间为3 h时,3a的收率高达80%,简化后处理;菲醌与溴素摩尔比为1.0∶3.4,反应时间为3 h时,3b的收率高达90%.再增加溴素与9,10-菲醌的摩尔比对目标产物的产率几乎无影响.另外,延长反应时间,目标产物的产率并未提高,但会使产品颜色加深.

图3 溴素与9,10-菲醌的物质的量的比值与收率的关系Fig.3 The relationship between the molar ratio of bromide to 9,10-phenanthrenequinone and their yields
3结语
9,10-菲醌与溴化剂进行选择性溴化反应,分别获得了较高收率的单溴代和双溴代产物.确定了单溴代反应的最优工艺条件为:用NBS做溴化剂时,9,10-菲醌与NBS的摩尔比为1.0∶1.3,在浓硫酸中0 ℃下反应3 h得到了2-溴-9,10-菲醌,收率60%;用溴素做溴化剂时,9,10-菲醌与溴素的摩尔比为1. 0∶1.4,用BPO做引发剂,在冰乙酸中106 ℃反应3 h得到了3-溴-9,10-菲醌,收率85%.双溴代反应的最优工艺条件为:用NBS做溴化剂时,9,10-菲醌与NBS的摩尔比为1.0∶2.3,在浓硫酸中室温下反应3 h得2,7-二溴-9,10-菲醌,收率76%;用溴素做溴化剂时,9,10-菲醌与溴素的摩尔比为1.0∶3.4,在冰乙酸中106 ℃反应3 h得3,6-二溴-9,10-菲醌,收率90%.
参考文献
[1]郭建忠, 薛永强. 菲醌的合成及利用[J]. 太原理工大学学报, 2001, 32(2): 140-143.
[2]龚山华. 菲醌合成的绿色工艺研究[D]. 太原:太原理工大学,2008.
[3]Muddasir H,Ping L,Mao L,et al.Synthesis,characterization,electrochemistry and optical properties of a novel phenanthrenequinonealt-dialkyl uorene conjugated copolymer[J]. Polym Int ,2007,56(12): 1507-1513.
[4]王志明. 菲醌的化学及衍生物的光电功能[D]. 长春: 吉林大学, 2011.
[5]Saito M, Yamamoto T, Osaka I, et al. Facile synthesis of [1]benzothieno [3,2-b]benzothiophene fromo-dihalostilbenes [J]. Tetrahedron Lett, 2010, 51(40): 5277-5280.
[6]Chatterjee T, Sarma M, Ghanta S, et al. Sterically driven electronic properties of naphthalene-and anthracene-end-capped 2,2′-bipyridine luminophores: synthesis and density functional theory[J]. Tetrahedron Lett, 2011, 52(42): 5460-5463.
[7]Song Y, Xu W, Zhu D. Synthesis and properties of cyclic ethylene-bridged 3,6-fluorene dimer and its linear analogues [J]. Tetrahedron Lett, 2010, 51(37): 4894-4897.
[8]Unver E K, Tarkuc S, Udum Y A, et al. The effect of the donor unit on the optical properties of polymers [J]. Org Electron, 2011, 12(10): 1625-1631.
[9]Pascal L, Eynde J J V, Haverbeke Y V, et al. Synthesis and characterization of novelpara-andmeta-phenylenevinylene derivatives: fine tuning of the electronic and optical properties of conjugated materials [J]. J Phys Chem B, 2002, 106(25): 6442-6450.
[10]祷新德, 丘坤元.Wohl-Ziegler:反应的研究芳香醚的侧链溴化与苯环溴化[J]. 化学学报,1959,25(5): 277-287.
[11]Masahiro N, Kazuki N, Eigo M, et al. Isomerically pure anthra [2,3-b: 6, 7-b′]-difuran (anti-ADF),-dithiophene (anti-ADT), and-diselenophene (anti-ADS): Selective synthesis, electronic structures,and application to organic field-effect transistors[J].J Org Chem, 2012, 77(18): 8099-8111.
Selective Bromination of 9,10 Phenanthrenequinone
YangHanmin1,ShenQianyan1,LiuChangyu1,ZhengWen2,TaoJun2
(1 College of Chemistry and Material Science, South-Central University for Nationalities,Wuhan 430074,China;2 Wuhan Pengo Technology. LTD, Wuhan 430074,China)
AbstractA series of 9,10-phenanthrenequinone bromides were selectively synthesized by varying reaction conditions such as the molar ratio of reactants, the type of solvent and the reaction temperature. 2-bromine-9,10-phenanthrenequinone was prepared by mixing 9,10-phednanthrenequinone withN-bromobutanimide in a molar ratio of 1.0∶1.4 at 0 ℃ for 3 h with concentrated sulfuric acid as solvent, and a 60% yield was obtained. Under similar condition, 76% yield of 2,7-dibromo-9,10- phenanthrenequinone was obtained with a molar ratio of 9,10-phenanthrenequinone toN-bromobutanimide of 1.0∶2.3 at room temperature.85% yield of 3-bromine-9,10-phenanthrenequinone was prepared by mixing 9,10-phednanthrenequinone and bromine in a molar ratio of 1.0∶1.4 at 106 ℃ for 3 h with acetic acid as solvent, and benzoyl peroxide as initiating agent. 90% yield of 3,6-dibromo-9,10-phenanthrenequinone was obtained with the molar ratio of 9,10-phenanthrenequinone to bromine of 1.0∶3.4 at the same condition. The selective bromination of 9,10-phenanthrenequinone was discussed and it was found that the amount of brominating reagent was the major factor determining the production of monobromide or dibromide.
Keywords9,10-phenanthrenequinone; bromo-derivation; bromination reaction; brominating agent; selective
收稿日期2015-09-14
作者简介杨汉民(1968-), 教授, 博士, 研究方向: 贵金属纳米催化剂的制备, E-mail: yanghanmin@sina.com
基金项目国家自然科学基金资助项目( 21203253)
中图分类号TQ 594
文献标识码A
文章编号1672-4321(2016)02-0015-04
猜你喜欢
煤矿安全(2022年9期)2022-09-16
城市道桥与防洪(2022年3期)2022-05-08
辽宁农业科学(2021年4期)2021-09-03
安全与环境工程(2021年2期)2021-04-02
陶瓷学报(2020年6期)2021-01-26
煤炭加工与综合利用(2020年6期)2020-07-17
世界农药(2019年3期)2019-09-10
中学生数理化·高二版(2016年3期)2016-12-26
安徽医科大学学报(2015年9期)2015-12-16
郑州大学学报(工学版)(2015年1期)2015-03-24
