
不同养殖模式下凡纳滨对虾肠道及环境微生物群落结构分析与比较
2023-12-11 10:06张建雄马瑞欣吴江爱崔亚楠郑百芹张丽芳
河南农业科学 2023年11期
侯 蔷,周 鑫,张建雄,马瑞欣,吴江爱,崔亚楠,陈 宇,郑百芹,张丽芳
(1.唐山市食品药品综合检验检测中心/农业农村部农产品品质评价与营养健康重点实验室,河北 唐山 063000;2.河北省水产技术推广总站,河北 石家庄 050000)
凡纳滨对虾(Litopenaeus vannamei)俗称南美白对虾,原种主要分布于太平洋沿岸热带水域,于1988 年引入我国。由于具有环境适应能力强、成活率高、生长速度快、出肉率高等优点,凡纳滨对虾目前在全国沿海省市均有养殖,养殖面积以及产量已占水产养殖业主导地位[1]。2020 年,全国凡纳滨对虾养殖产量达186万t以上,其中河北省凡纳滨对虾养殖产量达4.1 万t,且淡水养殖的对虾产量连年高于海水养殖[2]。作为北方地区的主要产区之一,河北省唐山市凡纳滨对虾的养殖面积逐年增大,养殖模式也由传统的海水粗放养殖模式,逐渐改进为精养、混养及工厂化养殖等高密度集约化养殖模式。
尽管养殖规模逐年扩大,但仍然存在较多因素制约着凡纳滨对虾养殖产业的健康发展,如细菌或病毒感染引发的病害等[3-4]。前人研究表明,虾类疾病与虾肠道及周围环境的微生物群落结构密切相关[5],肠道微生物群落的时序性变化与对虾疾病的严重程度呈正相关[6],如对虾白粪综合征(White feces syndrome,WFS)等;而养殖环境微生物紊乱则可能改变肠道微生物群落结构,进而抑制对虾生长甚至诱发疾病[7],如出现红肠、空肠等症状的患病对虾,其养殖塘水及肠道中变形菌门(Proteobacteria)和放线菌门(Actinobacteria)相对丰度均显著低于健康对虾。因此,研究对虾肠道和周围环境微生物的组成及其相互关系,对凡纳滨对虾健康养殖具有重要意义。目前,针对凡纳滨对虾肠道及周围环境微生物群落结构的研究主要集中在我国南方地区,如海南[5]、浙江[8-9]、广东[10-11]、上海[12]等省市,研究内容主要涉及凡纳滨对虾养殖过程中其肠道和养殖环境微生物群落的结构与变化,以及不同养殖模式或对虾不同生长状态下的微生物群落结构的比较等方面。然而,对于北方地区养殖的凡纳滨对虾则鲜有此方面的研究,更多是关注养殖水域微生物群落的结构及多样性,如罗同阳等[13]系统分析了白洋淀水域的细菌多样性。综上所述,目前,尚无关于我国北方特别是河北省唐山市凡纳滨对虾肠道及养殖环境微生物群落结构的研究。为此,选取唐山市2 种凡纳滨对虾养殖模式淡水精养模式(Freshwater intensive culture,FI)和混养模式(Freshwater mixed culture,FM),利用Illumina HiSeq 平台,对2 种模式下不同养殖时期采集的对虾肠道、养殖塘水体和底泥样品中微生物的16S rRNA 基因V3-V4 可变区进行高通量测序,分析比较各组样品中微生物群落结构及差异,探究唐山市养殖凡纳滨对虾微生物群落的时序性变化,为科学制定对虾养殖规划、优化养殖模式、建立健康养殖系统、提高养殖技术水平提供参考。
1 材料和方法
1.1 材料与仪器
凡纳滨对虾、养殖塘水体及底泥样品均采集自河北省唐山市某对虾养殖场;MagPure DNA Stool LQ Kit(广州美基生物科技有限公司产品);Qubit®dsDNA BR Assay Kit(美 国Invitrogen 公 司);2×Phanta Max Master Mix(南京诺唯赞生物科技有限公司);Agencourt AMPure XP 磁 珠(美 国Beckman Coulter公司);琼脂糖(西班牙Biowest公司)。
JC-801A 活塞式柱状采泥器(青岛聚创环保集团有限公司);LYNX4000 高速离心机(美国ThermoFisher 公司);水平电泳仪及凝胶成像系统(北京六一生物科技有限公司);T100™PCR 扩增仪(美国Bio-rad 公司);Agilent 2100 Bioanalyzer(美国Agilent 公司);Illumina Miseq 测序仪(美国Illumina公司)。
1.2 方法
1.2.1 样品采集地点及时间 样品采集地点位于河北省唐山市落潮湾某凡纳滨对虾养殖场(北纬39°13′53″,东经118°19′14″)。该养殖场同时拥有淡水精养(FI)和淡水混养(FM)模式养殖塘,每个养殖塘面积约26 000 m2,均为长方形,平均水深为1.9 m。FI模式养殖塘仅投放凡纳滨对虾,放养密度为60 尾/m2;FM 模式养殖塘凡纳滨对虾放养密度为22.5 尾/m2,同时套养草鱼(2.4 尾/m2)、花鲢(0.4尾/m2)和白鲢(0.4 尾/ m2)。养殖过程中均不换水,适当加水,每日投喂2 次。选取FI 模式和FM 模式养殖塘各1 个,分别于养殖初期(养殖第10 天)和养殖末期(养殖第120 天)采集凡纳滨对虾肠道、养殖塘水体及底泥样品。养殖初期采样时平均水温为21.5 ℃;养殖末期采样时平均水温为25.3 ℃。
1.2.2 样品采集方法及处理 每个时间点、每种模式养殖塘按照如下方式进行样品采集及后续处理。对虾肠道样品:随机捕捉24 尾凡纳滨对虾,低温保存带回实验室后,先用无菌生理盐水擦拭体表,随后在无菌环境下挑取肠道,每8 尾虾肠道合并作为1 个样品置于无菌离心管中,共3 个重复样品。水体样品:采用5 点采样法进行样品采集,养殖塘4 个角的采样位点均选在距离养殖塘岸边1.0~1.5 m 处的位置,连同养殖塘中点在内所有的采样位点均需远离食台、增氧机及进/排水口[12],分别在各个采样位点采集水面下25~35 cm 处的养殖塘水250 mL 于集水瓶中,低温保存带回实验室;混合后每取500 mL水为1 个样品,采用孔径为0.22 μm 的PVDF 膜过滤,收集滤液于无菌离心管中,共设置3 个重复样品。底泥样品:底泥样品采样位点的设置与水体样品一致;使用柱状采泥器分别在各个采样位点采集养殖塘底泥,切取柱状泥样前10 cm 的底泥于无菌采样袋,低温保存带回实验室;混合后分装置于3个无菌离心管中作为3 个重复样品。所有样品于-80 ℃条件下冻存。共采集36 个样品,2 种养殖模式各18个,各样品信息及编号详见表1和表2。
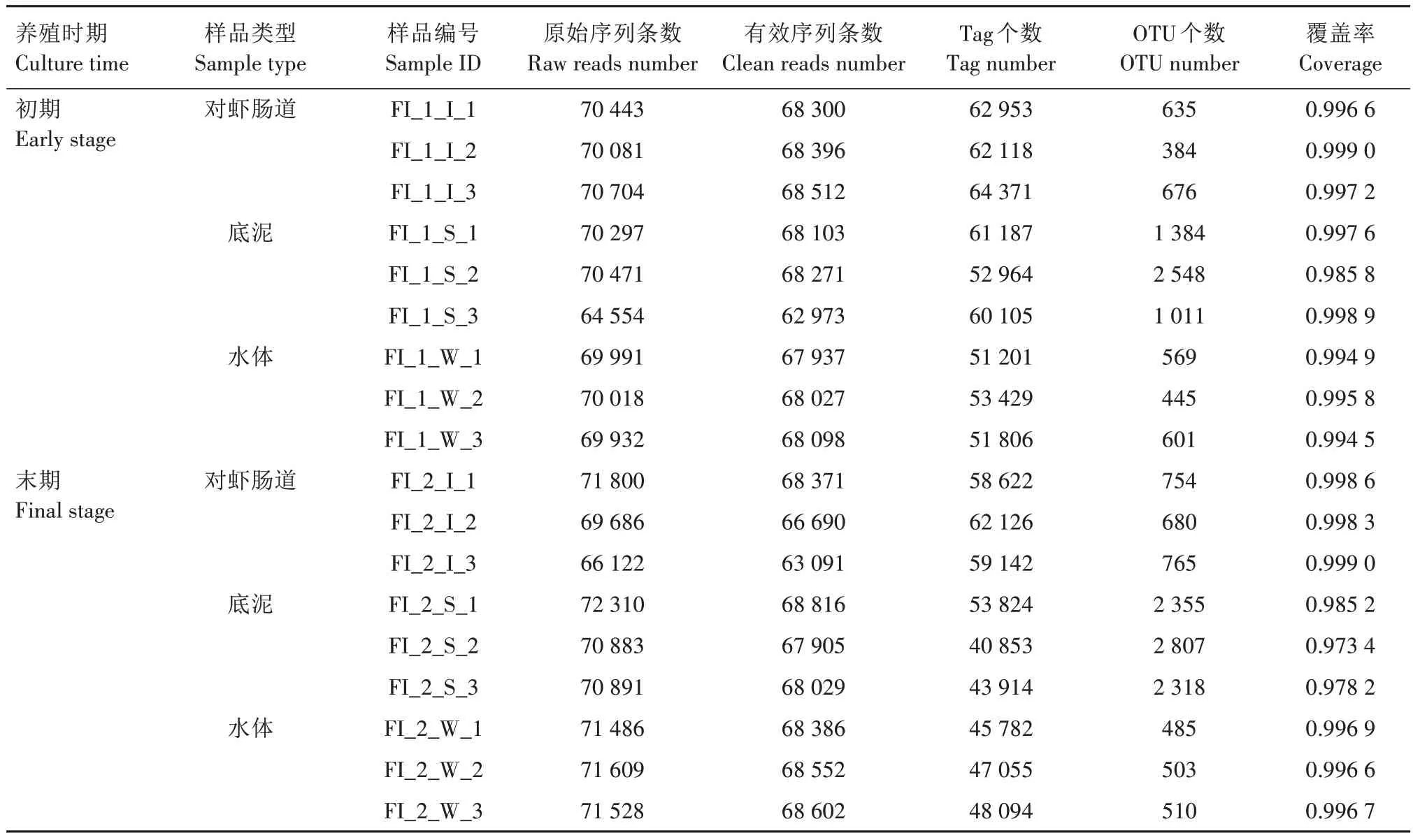
表1 淡水精养模式各组样品信息及高通量测序数据Tab.1 Statistical table of sample information and high-throughput sequencing data from FI pattern
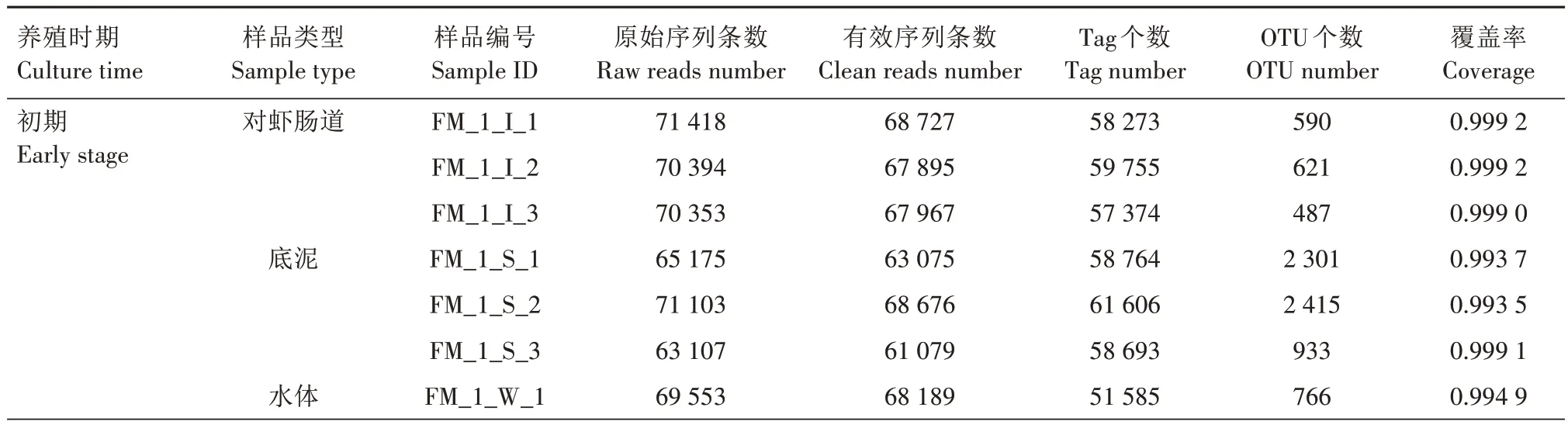
表2 淡水混养模式各组样品信息及高通量测序数据Tab.2 Statistical table of sample information and high-throughput sequencing data from FM pattern
1.2.3 DNA 提取与PCR 扩增 按照DNA 提取试剂盒说明书提取所有试验样品的总DNA,分别使用荧光定量法和1%琼脂糖凝胶电泳法检测DNA的浓度和完整性。取质量合格的DNA 作为模板,使用引物338F (5′-ACTCCTACGGGAGGCAGCAG-3′) 和806R(5′-GGACTACHVGGGTWTCTAAT-3′)对细菌16S rDNA V3-V4 区域进行PCR 扩增。扩增体系(50 μL):2×Phanta Max Master Mix 25 μL,引物338F和806R 各2 μL,DNA 模 板30 ng,ddH2O 补 足 至50 μL。扩增程序:94 ℃预变性3 min;94 ℃变性30 s,55 ℃退火45 s,72 ℃延伸45 s,30个循环;72 ℃终延伸10 min。
1.2.4 高通量测序及数据过滤 使用Agencourt AMPure XP 磁珠对PCR 扩增产物进行纯化,完成建库。使用Agilent 2100 Bioanalyzer 对文库的片段范围及浓度进行检测。检测合格的文库采用HiSeq 2500 进行高通量测序(深圳华大基因股份有限公司)。原始测序数据的过滤采取按窗口去低质量的方法,设置25 bp 的窗口,如果窗口平均质量值低于20 bp,从窗口开始截除reads 末端序列,移除最终reads 长度低于原始reads 长度75%的序列,去除接头污染的reads、含N 的reads和低复杂度reads,获得Clean data。序列拼接使用FLASH 软件[14],利用重叠关系将双末端测序得到的成对reads 组装成1条序列,得到高变区的Tag。
1.3 数据处理
获得的Clean tag 利用软件USEARCH 聚类为可操作分类单元(Operational taxonomic unit,OTU),即利用UPARSE[15]在97 %相似度下进行聚类,得到OTU 的代表序列,同时利用UCHIME[16]去除PCR 扩增产生的嵌合体。随后通过软件RDP classifer 将OTU 代表序列与Greengene[17]数据库比对进行物种注释,置信度阈值设置为0.6。根据每个样品的OTU 丰度计算独有及共有的OTU,通过R 语言工具包制作Venn 图及物种组成柱形图。利用软件Mothur[18]计算每个样品的Alpha 多样性指数,包括Chao 指数、Ace 指数、Shannon 指数和Simpson 指数,通过R 语言绘制Alpha 多样性组间差异盒型图。使用QIIME 软件[19],采用迭代算法在加权物种分类丰度信息的情况下,使用所有样品中序列数最少的样品的序列数的75%进行抽样分析,迭代100 次之后综合统计得到主坐标分析(Principal co-ordinates analysis,PCoA)展示图。利用软件phytools 及R 语言,根据Weighted Unifrac 距离矩阵绘制UPGMA 聚类分析图[20]。采用线性判别分析(LEfSe,https://huttenhower.sph.harvard.edu/galaxy/)比较不同样品微生物群落差异,确认主要特异类群。样本指数的显著性差异分析采用统计学t检验的方法进行。
2 结果与分析
2.1 高通量测序数据统计
根据试验设计方案所采集的36个对虾肠道、水体和底泥样品,通过16S rRNA基因测序获得的高通量数据如表1 和表2 所示。原始测序数据经过滤去除低质量序列及模糊序列,共获得有效序列条数为60 717~69 068;再经去除无法聚类和没有注释的序列后,拼接获得Tag 个数为40 853~64 371;以97%的相似水平进行OTU聚类,所获得样品的OTU个数为264~4 298。所有样品的测序覆盖率均大于0.972 2(0.972 2~0.999 4),表明此次测序数据能够代表样品中绝大多数细菌类群组成的真实情况,可进行后续的生物信息学分析。
同组间不同样品共有及独有OTU 数经加和后,得到不同养殖模式下各组样品OTU 数(图1a),绘制Venn图(图1b—d),分析比较不同养殖模式、不同养殖时间对虾肠道、水体和底泥样品中的微生物物种组成相似性及差异情况。FI 模式所有样品共聚类为6 077个OTU,FM模式所有样品共聚类为7 961个OTU,相较于FI 模式,FM 模式各组样品的物种数量更多。不同养殖模式的同类型样品中,对虾肠道包含125 个共有OTU,水体包含131 个共有OTU,底泥包含1 047 个共有OTU。总体而言,2 种养殖模式无论是在养殖初期还是末期,底泥样品聚类得到的OTU 数均为最高,显著高于对虾肠道和水体样品(P<0.05),且随养殖时间的延长而有明显增长,表明底泥样品中微生物数量最为丰富。
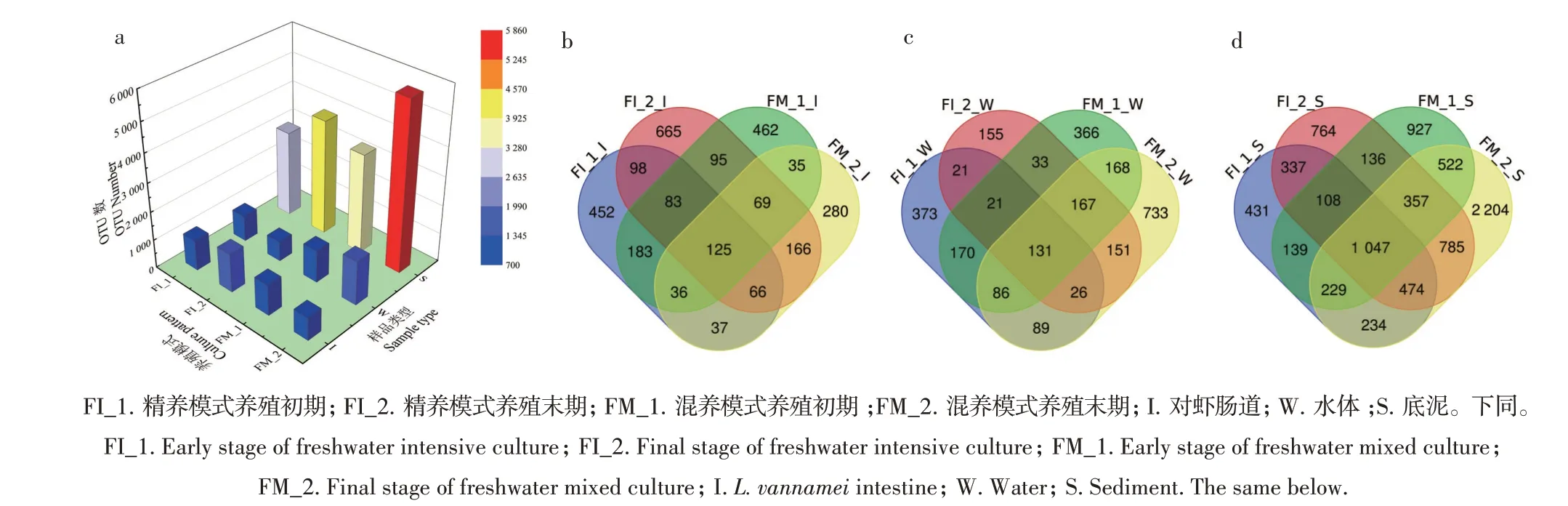
图1 不同养殖模式凡纳滨对虾肠道、底泥和水体样品OTU数统计(a)及Venn图(b—d)Fig.1 The counts(a)and Venn diagrams(b—d)of OTUs in L.vannamei intestine,sediment and water samples from different culture patterns
2.2 不同养殖模式下凡纳滨对虾肠道及养殖环境微生物多样性分析
2.2.1 Alpha 多样性分析 基于高通量测序结果对各组样品进行Alpha 多样性分析,得到多样性指数统计表(表3)及组间差异分析图(图2)。Chao 指数和Ace 指数可反映样品中物种的数量,而不考虑每个物种的丰度情况;Shannon 指数和Simpson 指数则反映样品的多样性受物种丰度和均匀度的影响,即相同物种丰度的情况下,样品中各物种具有越大的均匀度,则认为其具有越大的多样性[21]。由表3 可知,无论是FI 模式还是FM 模式,底泥样品的Alpha多样性均高于水体样品和对虾肠道样品;2 种养殖模式相比,FM 模式底泥样品的多样性更高。以各组样品Shannon 指数绘制组间差异分析图,如图2所示。对不同养殖时期采集样品的多样性进行比较可知,FI 模式养殖末期对虾肠道、底泥及水体样品多样性均高于养殖初期时相应样品(图2a);FM模式养殖末期底泥和水体样品多样性高于养殖初期,而对虾肠道样品多样性则低于养殖初期(图2b);养殖末期,FM 模式与FI模式相比,底泥样品及水体样品多样性更高,而对虾肠道样品多样性较低(图2c)。

图2 不同养殖模式凡纳滨对虾肠道、底泥和水体样品Shannon指数差异分析Fig.2 The differentiation analysis of Shannon index of L.vannamei intestine,sediment and water samples from different culture patterns

表3 不同养殖模式下各组样品Alpha多样性Tab.3 Alpha diversity statistics of samples from different culture patterns
2.2.2 Beta 多样性分析 对不同养殖模式、不同养殖时期各组样品中微生物群落进行PCoA 分析,结果如图3 所示,第一主坐标轴对样本差异的贡献度为26.37%,第二主坐标轴对样本差异的贡献度为20.34%。不同类型的样品微生物群落分区明显,其中底泥与对虾肠道微生物分区距离较近,表明底泥与对虾肠道的微生物组成结构相似度较高,群落差异较小;而水体和底泥、对虾肠道微生物组成均有差异。FI 模式下,不同养殖时期的底泥和水体样品分别聚类,群落相似性较高,而对虾肠道样品养殖末期与养殖初期分别聚类,微生物组成差异较大;FM 模式下,不同养殖时间的同类型样品聚集在一起,表明其微生物群落相似度较高,组成差异较小。
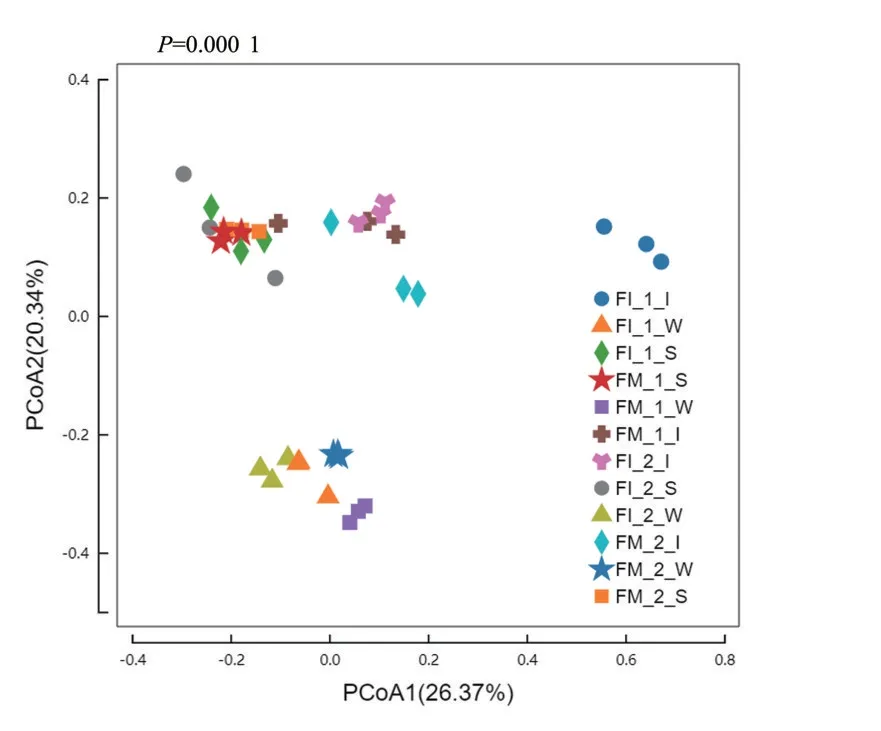
图3 不同养殖模式凡纳滨对虾肠道、底泥和水体样品PCoA聚类分析Fig.3 The PCoA cluster analysis of L.vannamei intestine,sediment and water samples from different culture patterns
2.3 不同养殖模式下凡纳滨对虾肠道及养殖环境微生物群落结构分析
2.3.1 基于门水平的微生物群落结构 在门水平上,2 种养殖模式、不同养殖时期的对虾肠道、底泥和水体样品的OTU 共注释到69 个门,汇总分析各组样品中相对丰度大于1%的物种,结果如图4 所示。FI 模式养殖初期,对虾肠道可注释到34 个门,其中蓝细菌门(Cyanobacteria,83.45%)、变形菌门(11.43%)和拟杆菌门(Bacteroidetes,3.01%)3 个门的相对丰度大于1%;水体样品可注释到39个门,其中放线菌门(61.07%)、变形菌门(21.65%)、拟杆菌门(8.39%)等5 个门的相对丰度大于1%;底泥样品可注释到56 个门,其中9 个门的相对丰度大于1%,包括变形菌门(41.61%)、拟杆菌门(13.08%)、放线菌门(11.95%)等。养殖末期,对虾肠道样品可注释到39 个门,其中8 个门的相对丰度大于1%,包括变形菌门(56.58%)、蓝细菌门(12.58%)、柔壁菌门(Tenericutes,8.42%)等;水体样品可注释到27 个门,包括放线菌门(45.95%)、拟杆菌门(18.93%)、变形菌门(15.46%)等在内的8 个门的相对丰度大于1%;底泥样品可注释到59 个门,其中变形菌门(38.84%)、放线菌门(16.38%)、拟杆菌门(15.07%)等8 个门的相对丰度大于1%。FM 模式养殖初期,对虾肠道样品可注释到36 个门,其中7 个门的相对丰度大于1%,包括变形菌门(57.88%)、蓝细菌门(16.46%)、拟杆菌门(12.6%)等;水体样品可注释到35 个门,其中8 个门的相对丰度大于1%,包括放线菌门(72.62%)、变形菌门(9.46%)、蓝细菌门(7.32%)等;底泥样品共注释到56个门,其中变形菌门(35.04%)、拟杆菌门(16.43%)、放线菌门(9.27%)等12 个门的相对丰度大于1%。养殖末期,对虾肠道样品共注释到33个门,其中柔壁菌门(60.7%)、变形菌门(20.11%)、蓝细菌门(7.28%)等7 个门的相对丰度大于1%;水体样品共注释到45 个门,其中9个门的相对丰度大于1%,包括放线菌门(23.87%)、蓝 细 菌 门(19.98%)、浮 霉 菌 门(Planctomycetes,15.92%)等;底泥样品共注释到65个门,包括变形菌门(35.01%)、拟 杆 菌 门(13.7%)、绿 弯 菌 门(Chloroflexi,10.33%)等在内的14 个门的相对丰度大于1%。总体来看,FM 模式下整体微生物群落中的细菌数量及相对丰度均更高。FI 和FM 2 种养殖模式不同养殖时期对虾肠道、水体和底泥样品中,优势门类均为变形菌门、拟杆菌门及放线菌门;相较于对虾肠道和水体样品,底泥样品中微生物门类的数量和丰富度均为最高。与养殖初期相比,养殖末期时各类型样品中微生物物种组成基本一致,但是物种群落相对丰度更高,FI 模式养殖末期疣微菌门(Verrucomicrobia)、厚壁菌门(Firmicutes)、绿菌门(Chlorobi)、浮霉菌门、梭杆菌门(Fusobacteria)、衣原体门(Chlamydiae)、WS6 等物种丰度均有显著提高(P<0.05);FM 模式养殖末期疣微菌门、绿弯菌门、浮霉菌门、柔壁菌门、梭杆菌门、螺旋体门(Spirochaetes)等门类相对丰度均有显著提高。
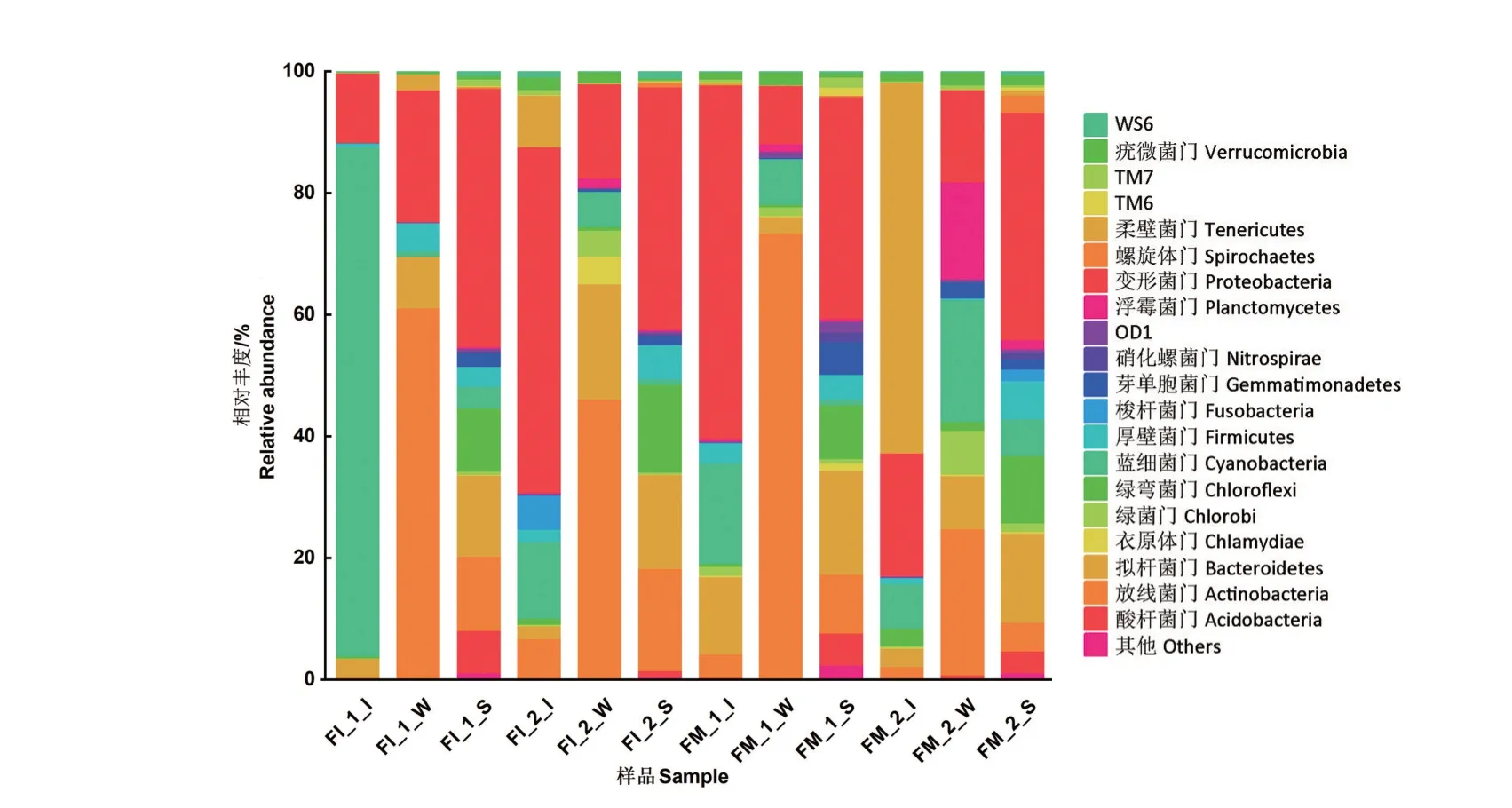
图4 不同养殖模式凡纳滨对虾肠道、底泥和水体样品基于门水平的微生物物种组成及相对丰度统计(>1%)Fig.4 Composition and relative abundance of microbiota of L.vannamei intestine,sediment and water samples from different culture patterns at phylum level(>1%)
2.3.2 基于属水平的微生物群落结构 在属水平上,2 种养殖模式、不同养殖时期的对虾肠道、底泥和水体样品的OTU 共注释到1 028 个属,其中35 个属的相对丰度大于1%,各组样品物种相对丰度汇总结果如图5所示。不同养殖模式下不同样品组的优势菌属组成及丰度明显不同。FI 模式养殖初期,对虾肠道样品主要优势菌属为红杆菌属(Rhodobacter,2.61%)和黄杆菌属(Flavobacterium,1.33%);水体样品主要优势菌属为Polynucleobacter(7.89%)、Limnohabitans(5.32%)、嗜 氢 菌 属(Hydrogenophaga,2.92%)等;底泥样品主要优势菌属为Gillisia(5.55%)、溶杆菌属(Lysobacter,5.38%)、硫杆菌属(Thiobacillus,3.05%)等。养殖末期时对虾肠道和底泥样品中属水平物种组成及丰度均有一定提升,对虾肠道主要优势菌属为弧菌属(Vibrio,16.52%)、希瓦氏菌属(Shewanella,8.13%)和鲸杆菌属(Cetobacterium,5.39%)等。底泥样品主要优势菌属 为Gillisia(5.62%)、海 杆 菌 属(Marinobacter,4.93%)、嗜盐单胞菌属(Halomonas,4.11%)等;水体样品主要优势菌属为Candidatus_Aquiluna(8.35%)、黄杆菌属(3.51%)、Candidatus_Xiphinematobacter(1.66%)等。FM 模式养殖初期,对虾肠道样品主要优势菌属为长命菌属(Rubrivivax,13.48%)、嗜氢菌属(8.66%)、黄杆菌属(8.2%)等;水体样品主要优势菌 属 为 聚 球 菌 属(Synechococcus,4.43%)和Candidatus_Xiphinematobacter(1.9%);底泥样品主要优势菌属为单胞菌属(Polaromonas,4.63%)、红育菌属(Rhodoferax,2.53%)、Lutibacter(2.5%)等。养殖末期,对虾肠道、水体及底泥样品共有的优势菌属为聚球藻属(Synechococcus),相对丰度分别为4.75%、11.56%和3.36%。此外,对虾肠道样品主要优势菌属还包括弧菌属(5.93%)、发光杆菌属(Photobacterium,3.25%)和希瓦氏菌属(1.71%);水体样品主要优势菌属还包括长命菌属(1.72%)和Candidatus_Xiphinematobacter(1.68%);底泥样品主要优势菌属还包括硫杆菌属(3.3%)、Lutibacter(1.37%)和脱硫球菌属(Desulfococcus,1.34%)。

图5 不同养殖模式凡纳滨对虾肠道、底泥和水体样品基于属水平的微生物物种组成及相对丰度统计(>1%)Fig.5 Composition and relative abundance of microbiota of L.vannamei intestine,sediment and water samples from different culture patterns at genus level(>1%)
2.4 不同养殖模式下凡纳滨对虾肠道及养殖环境微生物物种组成聚类分析
对2种养殖模式不同养殖时期的各组样品微生物群落的相似性进行聚类分析,绘制成UPGMA 图(图6),结果显示,微生物物种分别在不同类型样品(底泥、对虾肠道、水体)上进行聚类,而非以不同养殖模式或不同养殖时期进行聚类,表明不同类型的样品间物种组成差异更为显著。
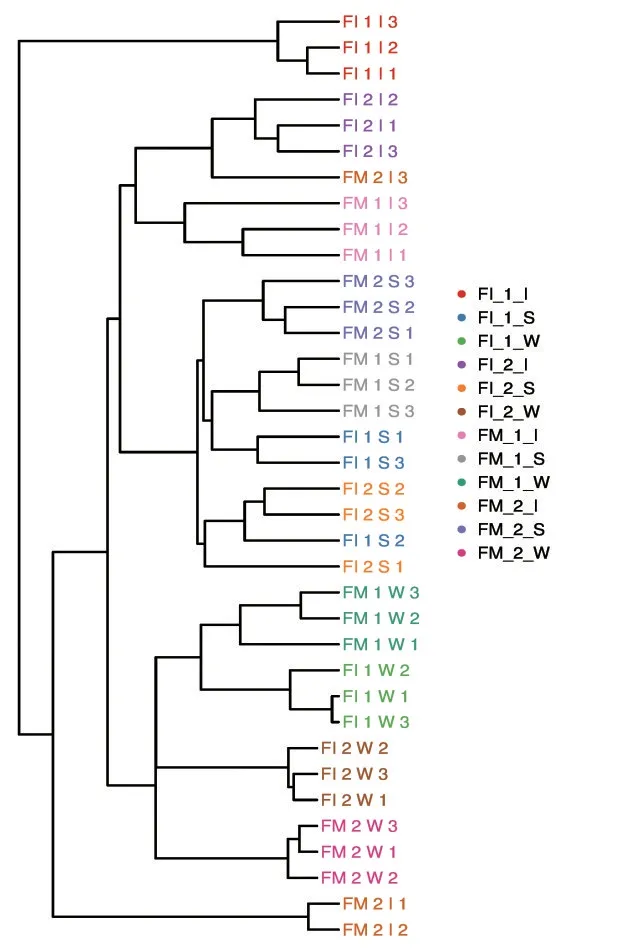
图6 不同养殖模式凡纳滨对虾肠道、底泥和水体样品UPGMA聚类分析Fig.6 UPGMA cluster diagram of L.vannamei intestine,sediment and water samples from different culture patterns
从图6 聚类分析上可以看出,不同类型样品的微生物群落结构有所不同,底泥与对虾肠道样品微生物群落结构较为相近,聚为一组;水体样品微生物群落结构则单独聚类为一组。热图分析横向聚类结果(图7)同样表明,底泥与对虾肠道样品微生物种类及组成较为相似,其相对丰度最高的物种均为变形菌门,其次为拟杆菌门;而水体样品微生物种类与其他2 组样品差异较大,其丰度最高的物种为放线菌门,其次为变形菌门。
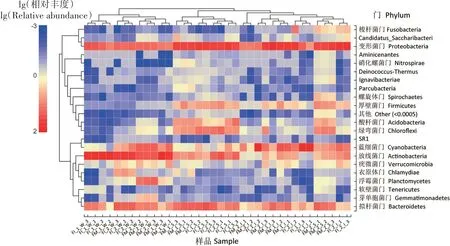
图7 不同养殖模式凡纳滨对虾肠道、底泥和水体样品门水平上微生物物种分类热图Fig.7 Taxa heatmap of microbiota of L.vannamei intestine,sediment and water samples from different culture patterns at phylum level
2.5 不同养殖模式下凡纳滨对虾肠道及养殖环境微生物指示物种线性判别分析
对各组样品间微生物群落差异进行线性判别分析,筛选组间主要的特异类群,确认对不同养殖模式、不同养殖时期微生物群落产生显著性影响的指示物种,结果如图8所示。养殖初期,FI模式下的指示物种在目水平上包括属于变形菌门的红杆菌目(Rhodobacterales)、根瘤菌目(Rhizobiales)和弯曲菌目(Campylobacterales),在科水平上包括属于变形菌门的伯克氏菌科(Burkholderiaceae)、嗜甲基菌科(Methylophilaceae)和螺杆菌科(Helicobacteraceae),放线菌门的微杆菌科(Microbacteriaceae)以及拟杆菌门的噬纤维菌科(Cytophagaceae);FM模式下的指示物种在目水平上包括属于变形菌门的假单胞菌目(Pseudomonadales)、拟杆菌门的噬纤维菌目(Cytophagales)以及放线菌门的酸微菌目(Acidimicrobiales),在科水平上包括属于变形菌门的鞘脂杆菌科(Sphingobacteriaceae)、丛毛单胞菌科(Comamonadaceae)和着色菌科(Chromatiaceae),拟杆菌门的黄杆菌科(Flavobacteriaceae),放线菌门的分支杆菌科(Mycobacteriaceae)以及厚壁菌门的毛螺菌科(Lachnospiraceae)。养殖末期时2 种养殖模式的特异类群在门水平上均有增加,相应的指示物种也发生了变化,如FI模式下的指示物种在门水平上增加了厚壁菌门和衣原体门,在纲水平上增加了属于厚壁菌门的芽孢杆菌纲(Bacilli),在目水平上变 为 属 于 变 形 菌 门 的 海 洋 螺 菌 目(Oceanospirillales)、放线菌门的土壤红杆菌目以及衣原体门的衣原体目(Chlamydiales),在科水平上变为属于变形菌门的希瓦氏菌科(Shewanellaceae)、军团 菌 科(Legionellaceae) 、甲 基 球 菌 科(Methylococcaceae)、脱硫杆菌科(Desulfobacteraceae)、交替单胞菌科(Alteromonadaceae)、盐硫杆状菌科(Halothiobacillaceae),放线菌门的间孢囊菌科(Intrasporangiaceae),拟杆菌门的噬几丁质菌科(Chitinophagaceae)以及厚壁菌门的梭菌科(Clostridiaceae_3);相较于FI 模式,FM 模式下的指示物种变化更为显著,在门水平上增加了芽单胞菌门(Gemmatimonadetes)、浮霉菌门、螺旋体门、酸杆菌门(Acidobacteria)以及柔壁菌门,在纲水平上增加了属于变形菌门的 δ - 变形菌纲(Deltaproteobacteria)、浮霉菌门的Phycisphaerae、酸杆菌门的全噬菌纲(Holophagae)以及柔壁菌门的柔膜菌纲(Mollicutes),在目水平上变为属于芽单胞菌门的芽单胞菌目(Gemmatimonadales)和螺旋体门的螺旋体目(Spirochaetales),在科水平上变为属于变形菌门的弧菌科(Vibrionaceae)和产碱菌科(Alcaligenaceae),厚壁菌门的单胞菌科(Syntrophomonadaceae)和 氨 基 酸 球 菌 科(Acidaminococcaceae)。
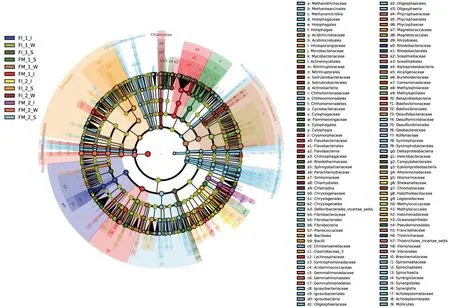
图8 不同养殖模式凡纳滨对虾肠道、底泥和水体样品微生物群落线性判别分析Fig.8 Linear discriminant analysis of microbiota of L.vannamei intestine,sediment and water samples from different culture patterns
3 结论与讨论
传统的微生物群落结构分析多采用变性梯度凝 胶 电 泳(Denatured gradient gel electrophoresis,DGGE)或末端限制性片段长度多态性(Terminal restriction fragment length polymorphism,T-RFLP)等[22-23]分子生物学技术,但这2 种技术分析通量较低,无法全面揭示复杂样品中的微生物组成及多样性。近年来,随着分子生物学技术的快速发展和数据分析方法的不断优化,高通量测序技术在微生物群落结构研究中获得了广泛应用[24]。本研究即采用Illumina HiSeq 高通量测序方法,对河北省唐山市淡水养殖FI 和FM 2 种模式下,凡纳滨对虾肠道及养殖环境样品微生物多样性和群落结构进行分析,研究显示,2种养殖模式无论是在养殖初期还是末期,底泥样品聚类得到的OTU 数最多,Alpha 多样性最高,明显高于对虾肠道和水体样品,且随养殖时间的延长而有明显增加,表明底泥样品中微生物群落最为丰富。这与金若晨等[12]对上海市凡纳滨对虾养殖的研究结论一致。此外,随着养殖时间的延长,FM 模式较FI 模式菌群相对丰度的变化更加显著,由于FI 模式只饲养凡纳滨对虾,养殖环境相对单一,而FM 模式是鱼虾混养,养殖环境受到影响的因素增多,因此微生物群落结构相对更加复杂。
水产养殖过程中,了解肠道及养殖环境的优势菌群,对养殖模式的选择和预防疾病暴发至关重要。在门水平上,作为淡水养殖系统中分布最广泛的两大细菌门类[25],变形菌门细菌广泛存在于土壤及动植物体内,其丰度的提高可作为人体肠道微生物组成失衡的标志[26],但其在对虾肠道菌群中的作用尚未得到深入研究[10];拟杆菌门细菌则是一个系统发育高度多样化的类群,主要在富营养化程度较低的湖泊中占优势,可将复杂分子(如多糖)加工转化成为简单化合物[27]。HE等[5]对海南省凡纳滨对虾肠道和养殖水体的微生物组成和功能进行研究,结果显示,所有样品中的优势门类均为变形菌门(38.70%~61.90%)和拟杆菌门(5.37%~25.11%);赵月季等[9]对浙江省不同养殖模式下凡纳滨对虾肠道核心微生物类群组成研究结果显示,变形菌门(61.1%)占据优势地位;ZHANG 等[28]通过16S rRNA测序和宏基因组测序2 种方法,研究了广东省凡纳滨对虾传统养殖模式下养殖环境的微生物群落,结果表明,变形菌门是所有样品中最丰富的门类;PEI等[29]研究发现,凡纳滨对虾肠道微生物的优势菌门为变形菌门、拟杆菌门和柔壁菌门,其中变形菌门是绝对优势门类(32.248%~32.40%),显著高于其他门类细菌;罗同阳等[13]对河北白洋淀水域细菌多样性的研究结果显示,变形菌门、拟杆菌门和放线菌门是湖水的优势菌门,且以变形菌门为绝对优势菌门(44.15%~60.18%)。本研究结果与上述研究结果基本一致,变形菌门是所有样品中相对丰度均较高的物种(9.46%~57.88%),其次为拟杆菌门(3.01%~18.93%)。此外,放线菌门在水体样品中丰度最高,绿弯菌门、厚壁菌门和酸杆菌门等在底泥样品中的丰度最高。放线菌门细菌是一类具有重大实用价值的微生物,能够代谢产生具有生物活性的产物,可用于分离筛选具有益生功能的菌株,用于水产养殖病害的预防与控制[30]。绿弯菌门细菌多定殖于海洋、潮间带及淡水的表层沉积物中,其可在无氧环境下进行光合作用[10]。绿弯菌门、酸杆菌门等门类细菌均具有氧化还原环境中过剩的有机物碳、氨氮和亚硝酸盐的能力,对于维持环境稳定起到重要的作用[31]。在属水平上,有研究表明,作为一种条件致病菌,弧菌属细菌是对虾养殖业中常见的病原微生物之一,其丰度的提高可能会影响对虾的健康状况甚至引起疾病[8]。因此,有必要监测养殖过程中弧菌属细菌的丰度变化,以降低对虾疾病暴发的风险[5]。本研究中,凡纳滨对虾养殖末期时,2种养殖模式下的对虾肠道中弧菌属相对丰度较养殖初期均有显著提高,由此推测养殖末期时的对虾具有更高的患病风险,在实际生产过程中应开展重点监测。
肠道及环境微生物在维持凡纳滨对虾的生长和健康方面有重要的作用[32-33],特别是肠道微生物,能够促进肠道消化吸收平衡、参与宿主免疫和代谢、维持宿主健康[34-37]。此外有研究表明,疾病会影响凡纳滨对虾肠道微生物的群落结构,健康对虾比患病对虾具有更高的细菌Alpha 多样性[7,38]。本研究中,FI 模式养殖末期凡纳滨对虾肠道及养殖环境样品微生物多样性均高于养殖初期时相应样品,而FM 模式对虾肠道样品微生物Alpha 多样性则随着养殖时间的变化呈现降低的趋势,且低于同时期FI模式的对虾肠道样品,结合养殖末期时FM 模式对虾肠道的指示物种包括弧菌科类群,提示本研究中FM 模式养殖末期时的凡纳滨对虾存在较高的患病风险。本研究每种养殖模式只选取了1 个养殖塘,结果存在一定的局限性。此外,因对虾肠道微生物群落的结构和功能还与环境因素息息相关[28],本研究中关于此方面尚有待于进一步研究。在实际生产中,有必要在对虾养殖期间随时关注环境因素的变化,特别是在FM 模式养殖的末期,以便及时调节养殖环境,避免因疾病暴发造成损失。
综上,本研究分析比较了河北省唐山市2 种淡水养殖模式的凡纳滨对虾肠道及养殖环境微生物群落结构,随着养殖时间的变化,对虾肠道、水体及底泥样品中微生物群落结构基本不变,优势物种均为变形菌门细菌,但是物种群落丰富度及多样性均有一定提高;2 种模式相比,FM 模式各个类型样品微生物群落物种数量更高,且养殖末期时物种相对丰度的变化更加显著;无论是FI还是FM 模式,底泥样品微生物群落的数量、丰度及多样性均为最高。通过本次研究,初步了解了唐山市淡水养殖对虾的微生态调控形式,为制定维持对虾健康的全菌群管理策略提供了基础信息,为促进凡纳滨对虾健康养殖及高品质对虾的生产提供了参考数据。
猜你喜欢
当代水产(2022年8期)2022-09-20
当代水产(2022年5期)2022-06-05
当代水产(2021年8期)2021-11-04
当代水产(2021年4期)2021-07-20
皮革制作与环保科技(2020年14期)2020-03-17
当代水产(2019年1期)2019-05-16
水科学与工程技术(2016年2期)2016-07-10
浙江大学学报(工学版)(2016年9期)2016-06-05
广东海洋大学学报(2015年4期)2016-01-13
铜业工程(2015年4期)2015-12-29
