
原发性干燥综合征合并神经系统病变患者32例临床分析
2024-02-02 12:17邹婵娟何善智丁菱王明霞王敏颜丝语
广东医学 2024年1期
邹婵娟, 何善智, 丁菱, 王明霞, 王敏, 颜丝语
中山市人民医院风湿免疫科(广东中山 528400)
原发性干燥综合征(primary Sjögren′s syndrome, pSS)是一种主要累及外分泌腺体的慢性自身炎症性疾病,除有唾液腺、泪腺功能受损而引起口干、眼干外,还可能引起多脏器的受累。临床中发现pSS合并神经病变并不少见,Teixeira等[1]报道,pSS神经系统并发症发生率为10%~60%。pSS神经系统病变早在20世纪40年代已有报道,可累及外周或中枢神经系统,部分pSS患者以神经系统损害为首发表现,严重影响患者的生活质量,pSS神经系统损害越来越受到重视,而目前缺少对pSS神经系统病变的诊断标准及共识,且关于神经系统病变的发病机制尚未明确。本研究拟回顾性分析我院收治的pSS合并神经系统病变患者的临床特征,旨在提高临床医师对pSS合并神经系统损害的认识,从而实现早期诊治、改善预后。
1 资料与方法
1.1 一般资料 2013年3月至2022年1月期间于中山市人民医院风湿免疫科和神经内科住院的pSS合并神经系统病变患者32例,所有患者均符合2002年pSS美国欧洲(US-EU)分类标准或2016年美国风湿病学会和欧洲抗风湿病联盟提出的pSS分类标准[2]。
排除标准:(1)重叠其他风湿性疾病;(2)同时合并心肌梗死、休克等危重疾病;(3)同时合并妊娠;(4)同时合并淋巴瘤、肺癌、卵巢癌、肠癌、甲状腺癌、胆囊癌、乳腺癌等恶性肿瘤。
本研究经本院临床科研和实验动物伦理委员会批准同意(2022-013号)。
1.2 临床资料收集 收集pSS合并神经系统损害患者的一般情况、临床表现、实验室指标、影像学资料、病理资料、治疗经过及转归,并根据欧洲抗风湿病联盟指定的干燥综合征疾病活动指数(EULAR Sjögren′s Syndrome Disease Activity Index, ESSDAI)[3]对患者病情活动进行评分。临床表现包括起病时间、首发症状、神经系统受累症状、口干眼干、关节损害、其他器官受累等;实验室指标包括血常规、生化、尿常规、抗核抗体(ANA)、ENA抗体谱、红细胞沉降率(ESR)、C反应蛋白(CRP)、免疫球蛋白、补体、脑脊液检查、水通道蛋白4(AQP4)抗体等;影像学资料包括头颅、脊髓影像学检查、神经电生理检查等。
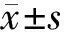
2 结果
2.1 一般情况 pSS合并神经系统损害患者共32例,占同期pSS住院患者的11.1%(32/287),其中女28例,男4例,女∶男为7∶1,年龄25~70岁,平均(51±12)岁。发病年龄(48.83±11.83)岁;病程6 h~360个月,中位病程1.50(0.15,37.25)个月。ESSDAI平均值为(14.38±8.61)分。
2.2 临床表现
2.2.1 pSS腺体受累 32例pSS合并神经系统损害的患者中,13例存在口干症状,10例存在眼干症状,20例行泪液分泌试验(Schirmer试验),15例阳性(Schirmer试验≤5 mm/5 min为阳性),其中10例双眼泪液分泌试验阳性,5例单眼泪液分泌试验阳性;29例患者同意完善唇腺活检,其中26例可见灶性淋巴细胞浸润,灶性指数均≥1灶/4 mm2,其中7例灶性指数为1灶/4 mm2,6例灶性指数为2灶/4 mm2,13例灶性指数≥3灶/4 mm2。
2.2.2 pSS腺体外表现 pSS合并神经系统损害的32例患者中,18例以神经系统表现为首发症状,占56.3%;24例(75%)以神经系统受累为唯一腺体外临床表现;余8例患者存在神经系统以外的全身其他系统受累表现,2例合并血液系统损害、肾小管酸中毒,1例合并肾小管酸中毒、肌肉病变,1例合并肾小管酸中毒,1例合并肌肉病变,1例合并肺间质病变,1例合并桥本氏甲状腺炎,1例合并关节病变。
2.2.3 pSS合并神经系统损害的临床表现 32例pSS合并神经系统损害的患者中,2例(6.3%)患者同时累及中枢神经系统与周围神经系统,27例(84.4%)单纯中枢神经系统受累,2例(6.3%)单纯周围神经系统受累,1例(3.1%)为神经-肌肉接头病变。
中枢神经系统受累患者共29例,占pSS合并神经系统损害患者的90.6%(29/32)。
其中,脑损害19例,占65.5%(含有16例单纯累及脑,2例同时累及脑及脊髓,1例同时累及脑及周围神经),4例表现为偏瘫,2例表现为构音障碍,1例表现为构音障碍及偏瘫,3例主要表现为反复发作的头痛,2例精神异常、烦躁、癫痫,2例症状不典型,表现为头晕,发作时间及间隔不定,1例表现为头痛、视物模糊,2例脑干受累,2例小脑受累。11例(37.9%)患者最终诊断脑血管疾病(脑梗死或短暂性脑缺血发作)。
脊髓损害共12例。其中单纯脊髓损害5例,5例均表现为急性横断性脊髓炎,主要表现为肢体乏力、肢体麻木、恶心呕吐、呃逆;视神经+脊髓损害者4例,均诊断为视神经脊髓炎谱系疾病(neuromyelitisoptica spectrum disorder, NMOSD),表现为急性横断性脊髓炎及视神经炎,主要表现为头痛、肢体乏力、视物模糊;脑、脊髓均损害者2例,诊断:视神经脊髓炎谱系疾病、急性播散性脑脊髓炎,表现为肢体麻木、头晕、呕吐、肢体乏力、失禁等;脊髓+周围神经受累1例,主要表现为急性横断性脊髓炎+周围神经病变。
值得注意的是,32例pSS合并神经系统损害患者中,9例诊断为NMOSD。NMOSD核心症状分型:视神经+脊髓3例,脊髓4例,脊髓+极后区1例,视神经+脊髓+极后区1例;视神经脊髓炎谱系疾病患者脊髓受累部位:颈段1例,胸段1例,颈段+胸段5例,颈段+胸段+延髓2例。
周围神经系统受累患者共4例,占pSS合并神经系统损害患者的12.5%(4/32)。其中2例累及周围神经(脊神经),最终诊断慢性炎性脱髓鞘性多发性神经根神经病,主要表现为肢体麻木、肢体乏力;1例累及脊髓及周围神经,最终诊断为中枢神经系统干燥综合征脊髓炎,主要症状为肢体麻木;1例累及脑桥及周围神经,诊断为脑桥梗死,周围神经受累主要症状为肢体麻木。
诊断神经肌肉接头病变者1例,占3.1%,该患者为46岁女性,因反复左侧眼睑下垂3年来诊,追问患者病史,患者起病时即伴有口干、眼干症状,但当时未予重视,来院后最终诊断为重症肌无力(眼肌型)。
32例患者ESSDAI评分分布主要为神经系统病变,其次为肾脏病变、肺部病变、肌肉病变、关节病变、血清学变化等,神经系统病变占比中位数(四分位数间距)为0.94 (0.52, 0.95)。
2.3 pSS合并神经系统损害的影像学检查结果 pSS合并神经系统损害的32例患者中,MRI检查异常者28例(87.5%)。在19例脑损害患者中,16例(84.2%)有MRI异常,其中基底节、放射冠区信号异常9例,额顶叶信号异常3例,颞叶信号异常2例,脑桥信号异常2例,小脑信号异常2例,侧脑室旁信号异常1例。12例脊髓病变患者中,长节段受累(≥3个节段受累)者9例,颈髓受累10例,胸髓受累10例,腰髓受累1例。
2.4 pSS合并神经系统损害的脑脊液检查结果 在32例pSS合并神经系统损害患者中,12例患者完善脑脊液检查,6例(50%)脑脊液结果异常,6例均有脑脊液蛋白增高,1例白细胞增多,以多个核细胞为主,2例脑脊液AQP4抗体阳性。
2.5 pSS合并神经系统损害的的免疫指标结果 32例pSS合并神经系统损害的患者中,ANA阳性率100% (29/29),抗SSA抗体阳性率93.8%(30/32),抗SSB抗体阳性率35.5%(11/31),高球蛋白血症的发生率为12.9%(4/31),低补体血症发生率为16.1%(5/31)。IgG 14.60(12.12,17.87)g/L,类风湿因子12.10(8.10, 21.70)IU/mL,ESR 34(25, 44)mm/h,CRP 2.75(0.60, 5.25)mg/L。7例患者血AQP4抗体阳性。
2.6 定性诊断 32例患者中,脱髓鞘性疾病13例[9例NMOSD、1例急性播散性脑脊髓炎、1例多发性硬化、2例慢性炎性脱髓鞘性多发性神经根神经病(慢性吉兰-巴雷综合征)],血管性疾病13例(脑梗死10例,短暂性脑缺血发作1例,脊髓动脉梗死2例),肌紧张性头痛2例,重症肌无力1例,感染性病变3例(病毒性脑炎2例,化脓性脑膜炎1例);
考虑到感染性病变、重症肌无力、肌紧张性头痛患者例数较少,未纳入统计,比较脱髓鞘性疾病、血管性疾病患者的临床指标,结果发现,脱髓鞘性疾病患者外周血淋巴细胞数量、血清IgM水平均高于血管性疾病患者[1.98(1.39,2.77)×109·L-1vs. 1.30(1.07,1.77)×109·L-1,P=0.005; 1.31(1.12,1.88)g/Lvs. 0.98(0.74,1.18)g/L,P=0.009];两者在年龄、性别、病程、炎性指标(ESR、CRP、RF)、生化指标、ESSDAI评分差异无统计学意义(P>0.05)。
2.7 pSS合并神经系统损害的治疗和转归 32例患者中,26例使用糖皮质激素,根据病情不同采用不同剂量,其中10例(31.3%)给予甲泼尼龙1 g/d冲击5 d,2例给予甲泼尼龙500 mg/d冲击5 d,1例予甲泼尼龙120 mg/d静脉滴注5 d,2例予地塞米松10 mg/d静脉推注5 d,2例予甲泼尼龙40 mg/d静脉滴注5~7 d,9例予口服泼尼松≤40 mg/d(含2例病毒性脑炎患者);其中3例联合免疫球蛋白冲击治疗,10例联合环磷酰胺,2例联合硫唑嘌呤,9例联合使用羟氯喹,1例联用甲氨蝶呤;1例单用羟氯喹,1例单用雷公藤,4例未予激素或免疫抑制治疗(1例合并感染,3例拒绝免疫抑制治疗)。32例患者中,29例好转或部分好转,3例无明显疗效,无死亡病例。
3 讨论
pSS是一种常侵犯外分泌腺体、B淋巴细胞异常增殖的慢性炎症性自身免疫病,主要表现为口干、眼干等症状,还可能出现腺体外表现,既往报道[4]提示pSS合并神经系统损害并不少见。本研究结果显示pSS合并神经系统病变的总体发生率为11.1%,与文献报道[1]的10%~60%发生率一致。不同研究报道的发生率差异较大,可能与缺乏统一的诊断标准、选择研究人群的偏倚有关[5]。

表1 pSS神经系统损害患者中脱髓鞘性疾病、血管性疾病患者临床特征比较 M(P25,P75)
本研究中,56.3%(18/32)的患者以神经系统表现为首发症状,75%(24/32)以神经系统受累为唯一腺体外临床表现,与相关报道[6-7]一致。ESSDAI是EULAR制定的用于评估干燥综合征病情活动的标准[3],法国一项395例pSS患者的前瞻性队列研究[8]发现,有神经系统受累者超过一半的ESSDAI由神经系统损害构成,而在本研究中,pSS合并神经系统病变患者ESSDAI评分神经系统病变占比更是高达0.94。因此,以神经系统为首发表现或唯一表现的患者,临床上容易忽略并延误诊断和治疗,有学者提出[6]不明原因出现神经系统病变的患者应注意考虑pSS。临床上需注意完善自身抗体谱、干眼症相关检查、唇腺活检等检查进一步排查pSS。
pSS神经系统病变临床表现多样,可累及中枢神经系统(CNS)、外周神经(PNS)。pSS合并CNS损害患者病变主要累及脑、脊髓及视神经。脑部病变包含局灶性和弥漫性病变,局灶性病变主要表现为局部感觉和运动异常、失语、癫痫发作、构音障碍和视觉减退等,弥漫性病变主要表现为亚急性或急性脑病、心理障碍、无菌性脑膜脑炎和认知障碍等[9]。本研究90.6%(29/32)pSS合并神经系统损害患者有CNS病变,以脑损害为主(19/29),其次为脊髓病变(12/29)、视神经病变(4/29)。pSS并CNS损害患者临床表现多种多样,本研究中最常见的临床表现为偏瘫、头痛、癫痫、构音障碍等,部分患者出现呃逆、行走不稳、失禁,脑损害患者中大部分(11/19)最终诊断为脑梗死或短暂性脑缺血发作,本研究中11例脑血管疾病患者中4例无高血压、糖尿病、高血脂、吸烟、肥胖以及长期激素治疗等脑血管病变的常见风险因素,有7例合并高血压(5例血压控制良好,2例血压控制欠佳),1例合并糖尿病(血糖控制良好),3例有吸烟史,考虑不能完全用传统风险因素解释脑血管病变,可能与pSS所致血管炎有关。既往研究认为,pSS血管损伤的病理机制是单核细胞炎症介导的缺血性或出血性脑血管炎,主要累及小血管,以中小静脉为主,也可累及小动脉,表现为炎性细胞浸润、血管壁纤维素样坏死、血管壁纤维化、动脉瘤形成或血管闭塞。在中枢神经系统,皮层下白质和脑室周围的血管最易受累,可表现为急性脑梗死、脑出血或慢性缺血所致的弥漫性缺血性脱髓鞘[5,10]。
本研究中pSS患者合并脊髓病变患者共12例,其中9例表现为急性横贯性脊髓炎,最终诊断为NMOSD,77.8%(7/9)患者抗AQP4抗体阳性。pSS合并NMOSD在临床中并不少见,协和医院一项研究[11]发现616例pSS患者中43例患者合并NMOSD。pSS与NMOSD均为自身免疫性疾病,两种疾病属于共病还是并发症关系还存在争议,有研究提出[12-13]“两者共病现象”的理论。NMOSD是一种免疫介导的以视神经和脊髓受累为主的中枢神经系统脱髓鞘疾病[14-15],其病因主要与抗AQP4抗体相关[16]。既往研究报道抗AQP4抗体滴度与患者病情活动度、病情复发均相关[17],在其他自身抗体、炎性因子等因素协同作用下抗 AQP4抗体更易致病,如破坏血脑屏障、产生抗AQP4抗体致病的环境[18]。
文献报道,pSS合并神经系统病变常见为周围神经病变[1],PNS病变以感觉型神经病最常见,临床多表现为肢体麻木疼痛、肌无力、末梢型感觉障碍等;也有学者报道了11例以神经系统病变为首发表现的pSS患者中3例诊断为急性吉兰-巴雷综合征,认为吉兰-巴雷综合征在pSS周围神经病变中并不少见[19];目前慢性炎性脱髓鞘性多发性神经根神经病合并pSS的病案报道罕见[20]。本研究中周围神经病变4例,2例诊断为慢性炎性脱髓鞘性多发性神经根神经病(两例患者脑脊液检查均存在蛋白细胞分离),2例诊断为多发性神经病。本研究中周围神经病变患者例数不多,可能与周围神经受累的患者多于门诊就诊有关,而本研究中选择的患者为住院患者。吉兰-巴雷综合征与pSS的关系如何尚需进一步观察及扩大病例数研究。
除CNS、PNS病变外,本研究中还发现1例pSS患者累及神经-肌肉接头,最终诊断重症肌无力。重症肌无力是以神经-肌肉接头损害为主的自身免疫性疾病,pSS合并重症肌无力极为少见,多为个案报道,Li等[21]报道了3例pSS合并重症肌无力患者,并行文献复习,共回顾分析了17例pSS合并重症肌无力患者的临床特征,结果表明pSS与重症肌无力并存的情况非常罕见,合并重症肌无力似乎不会对pSS病程产生不利影响。
目前pSS合并神经系统受累的发病机制未明,有研究指出中枢神经系统病变的机制可能与单核细胞浸润中枢、免疫介导血管损伤、小血管炎所致的组织缺血有关[22]。从定性诊断来看,本研究中pSS合并神经系统病变患者主要表现为脱髓鞘性疾病、血管性疾病,其次是感染性疾病、肌紧张性头痛,神经-肌肉接头病变最少见;脱髓鞘性疾病患者外周血淋巴细胞数量、血清IgM水平均高于血管性疾病患者,提示淋巴细胞活化、免疫系统激活与泛化可能在pSS神经系统受累的发病、疾病发展演变过程中发挥作用。
MRI检查对pSS合并神经系统损害的诊断帮助较大,既往报道pSS合并CNS受累患者MRI检查提示脑白质脱髓鞘病变(约70%)、脊髓长T2信号(75%)、皮层、小脑萎缩及脑室扩张等病变[23-24]。本研究发现MRI检查异常率达87.5%,其中脊髓异常信号12例,长节段受累(≥3个节段受累)9例,颈髓受累10例,胸髓受累10例,腰髓受累1例,与李伟等[25]报道的pSS脊髓病变构成基本一致。除脊髓异常信号、脑白质脱髓鞘病变外,脑缺血病变不少见,达34.3%(11/32),与李伟等[25]报道的pSS急性脑血管病发病率22.86%较接近。因此,临床上如怀疑pSS合并神经系统病变患者,应注意完善MRI检查进一步明确诊断。
目前pSS合并神经系统病变患者的治疗无明确的指南推荐,临床上以糖皮质激素联合免疫抑制剂为一线治疗方案。范薇等[26]研究显示对急性起病、症状明显或合并NMOSD的患者大剂量激素冲击治疗联合丙种球蛋白治疗及免疫抑制剂治疗是有效的,免疫抑制剂首选环磷酰胺,也有选择如甲氨蝶呤鞘内注射、利妥昔单抗、间充质干细胞等,患者症状得到不同程度的改善。不同神经系统病变类型可能发病机制不同,对免疫抑制剂的治疗疗效也存在差异。既往研究[22]提示激素对pSS相关的多发性神经病和多发性颅神经病有效,对感觉性共济失调效果不佳。稳定自限性患者可以不予特殊治疗,但应定期随访,注意有无复发。
综上所述,pSS合并神经系统病变并不少见。部分患者以神经系统病变为首发表现且为唯一腺体外表现,容易被医师忽略。患者神经系统受累类型多样复杂,脱髓鞘疾病、血管性疾病多见,发病机制不明确,淋巴细胞活化、免疫系统激活与泛化可能在发病、疾病演变过程中发挥作用。
利益相关声明:本文所有作者均声明无利益冲突。
作者贡献说明:研究设计为邹婵娟、何善智;研究方案执行与实施为邹婵娟、丁菱;数据整理为邹婵娟、王明霞、王敏、颜丝语;统计分析及论文撰写、投稿为邹婵娟。
猜你喜欢
中国实验诊断学(2023年5期)2023-05-26
昆明医科大学学报(2021年4期)2021-07-23
现代畜牧科技(2021年3期)2021-07-21
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际放射医学核医学杂志(2020年2期)2020-05-30
医学新知(2019年4期)2020-01-02
中国中医急症(2019年10期)2019-05-21
大众健康(2017年1期)2017-04-13
中国医科大学学报(2016年11期)2016-12-01
现代检验医学杂志(2016年4期)2016-11-15
