
增生性肌炎误诊软组织恶性肿瘤经验总结
2020-12-22 03:51罗光敏何贵生柯传庆彭恩兰
中国医药科学 2020年22期
罗光敏 何贵生 柯传庆 彭恩兰
1.贵州省黔西南州兴仁市人民医院内三科,贵州黔西南 562300;
2.解放军联勤保障部队第九〇八医院援助贵州专家组,江西南昌 330002
增生性肌炎(proliferative myositis,PM)是一种发生在肌肉组织的炎症性疾病,临床罕见,多见于中老年人,生长迅速,容易误诊为软组织恶性肿瘤,其实质属增生性良性病变,具有自限性,预后良好。我科2019年6月接诊1例由外院诊断为软组织肉瘤患者,转入后经细针穿刺细胞学检查确诊为PM,未行特殊治疗,发病5周后症状逐渐减轻,9周后包块基本消失,随访半年无异常,现报道如下。
1 临床资料
男,52岁,因左大腿包块半月入院。患者于2019年6月10日发现左大腿内侧包块,呈进行性肿大,轻微胀痛,运动时加重,无发热盗汗及其他不适,同月21日在某县医院诊治,CT平扫见左股骨内侧软组织占位,诊断为软组织恶性肿瘤,建议手术切除,2019年6月24日转入我院进一步诊治。原有高血压病史10年,无外伤史。查体:各浅表淋巴结未触及,心肺腹未及异常,左大腿内侧膝关节上方8cm处软组织明显隆起,颜色正常,皮温不高,可触及一软组织包块,大小7.5cm×5.5cm、质地中等偏硬、表面尚平、边缘欠清、固定不移动、轻压痛,左下肢运动无障碍,双下肢无浮肿。入院后查血常规及肝肾功能无异常,肿瘤标记物CEA、CA19-9、SCC、NSE均正常;MRI检查左股骨中间肌见片状T1异常信号影,STIR序列呈主信号影,范围7.0cm×5.2cm,边界不清,注射造影剂后呈明显强化,周围肌间隙亦可见高信号影,股骨正常(图1a、图1b);彩色超声见患处肌群局限性肿胀,大小6.2cm×3.9cm×3.2cm,内回声欠均匀,CDFI未见明显血流(图2),提示肌肉性病变;2019年6月26日细针穿刺细胞学检查见增生的纤维母细胞/肌纤维母细胞,部分可见小核仁,细胞分化尚可,提示良性纤维母细胞/肌纤维母细胞增生性疾病(图3)。经上级医院病理专家会诊,诊断:PM,排除软组织恶性肿瘤,建议取组织病理检查,患者拒绝,亦未行特殊治疗出院,2019年7月中旬症状逐渐减轻,包块缩小,2019年8月底包块基本消失,随访至2020年1月患者无异常。
2 讨论
PM是一种发生在肌组织的炎症性疾病,临床罕见,于1960年首次由Kern报道并命名,实质上是肌纤维周围的纤维结缔组织增生性炎症,并非肌纤维本身炎症,属良性病变,又称假肉瘤性良性病变,容易误诊为恶性肿瘤,具有自限性,预后良好。本文复习近年文献报告,综合分析其发病情况。
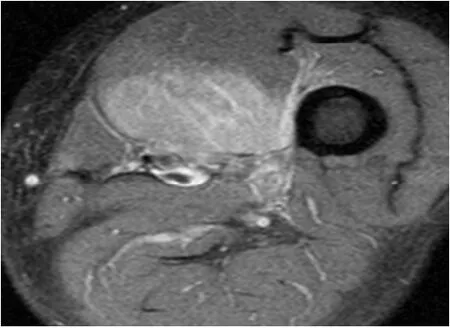
图1 a MRI横截面中间肌见片状T1异常信号,STIR序列呈主信号影,呈明显强化,周围肌间隙高信号影
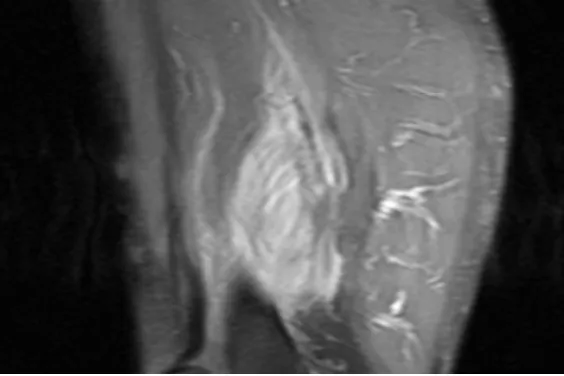
图1 b MRI冠状面
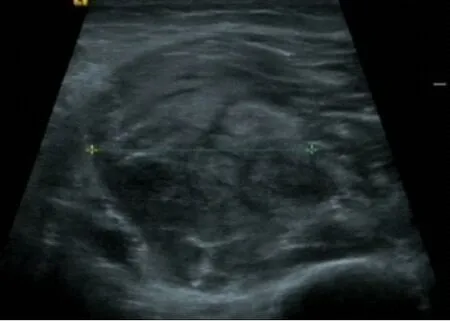
图2 彩色多谱勒显示内回声欠均匀,CDFI未见明显血流
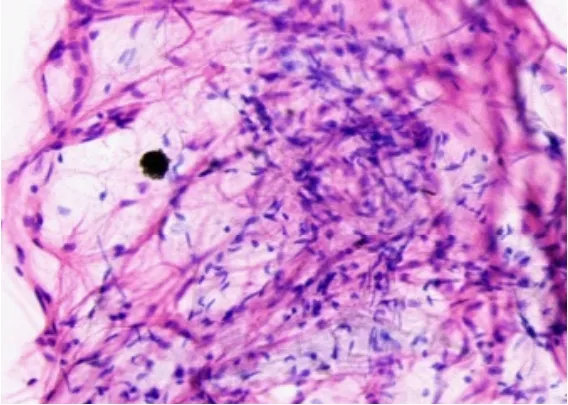
图3 细胞学可见增生的纤维母细胞/肌纤维母细胞,部分可见小核仁
2.1 临床表现
属罕见病,文献报告截至2016年世界范围内仅100余例[1],中老年多发,高峰在40~50岁,性别和种族差异不大[2],发病部位多在躯干、四肢,胸部和大腿最常见,生长迅速,病情变化快,局部软组织包块迅速增大,通常在数天至2周达最高,无畏寒、发热,部分患者可伴轻微胀痛,运动时加重,患处软组织呈块状隆起,皮温及颜色无异常,包块质地中等偏硬、表面平、边缘欠清、固定不移动、轻压痛,全身症状不明显或基本没有。
2.2 发病机制及病理生理
发病机制尚不明确,文献报告30%病例有外伤史,可能与局部损伤或肌肉缺血有关,局部缺血导致肌肉组织异常分化和生长[3-4],真正机制尚有待进一步探讨。病理生理主要以嗜碱性巨细胞及增生的成纤维细胞为病变特征,主要累及肌肉间质,而横纹肌细胞本身不受累,在横断面上可见“棋盘状”结构,局部肌肉组织呈不同程度萎缩,或完全被病变取代[5]。
2.3 实验室检查
无特异性,对诊断帮助意义不大,少数病例因炎症和疼痛刺激可导致白细胞和红细胞沉降率轻度增高,生化基本不受影响,肿瘤标志物大多阴性。
2.4 影像检查
首选MRI,可见包块位于肌肉层,沿肌肉方向生长,呈稍长T1长T2或混杂T2信号,界限不清,T1W1呈等信号,T2W1低信号网状影,周围可见水肿带,呈长T1长T2改变,注射造影剂后病灶明显不均匀强化,而内网状影不强化,边缘模糊,信号均匀,周围肌肉水肿,呈长T1长T2改变[6-7]。CT扫描包块呈高密度影,回声不均匀,呈典型“棋盘样”改变,其内可见坏死[8],由于其对软组织分辨率不及MRI,故较少用,在基层医院或MRI检查有禁忌时才选择。彩色超声是诊断PM又一重要方法,通常采用高频超声,其显示包块呈不均质中等偏高回声,可见粗大纤维,肌纤维内低回声,呈网格状,病灶成典型的“龟背样”“棋盘状”改变,CDFI多无血流信号,少数可有条索状少供血型信号,血流不丰富,且可记录到高阻动脉血流频谱[9-12],彩色超声操作简单方便,重复性好,还可引导穿刺活检;PET-CT检查可有FDG摄取轻度增高[13],一般不选择,对疑为恶性肿瘤者则有重要意义。
2.5 组织病理学和穿刺细胞学检查
是诊断PM最确切最重要的依据,组织病理学是金标准,典型表现镜下见肌肉间质内大量增生活跃的梭形细胞和成团的嗜碱性巨细胞,梭形细胞类似纤维母细胞,呈短梭形、圆形、或不规则形,核膜明显,染色质分布均匀,核空泡状,核仁清晰,细胞排列松散,呈束状或车辐状;嗜碱性巨细胞成群分布,类似神经节细胞,呈圆形或多边形,体积大,胞质丰富,核偏移;两种细胞常见核分裂,增生的细胞与肌肉分界不清,肌肉萎缩或被其他取代[1,7],其特征性表现一是纤维母细胞增生累及肌外膜、肌束和肌内膜,二是嗜碱性大细胞胞浆丰富,胞核呈空泡样,核仁明显;2013版WHO软组织和骨肿瘤分类将其归纳为纤维母细胞/肌纤维母细胞肿瘤(良性组)[14]。免疫组织化学,Vimnfin(+)、Aetin(+)、SMA(+)、Fibronectin(+)、Desmin 弱(+)、CD68(+)、Myosin 弱(+)、AACT(-)、Cytokeratin(-)、S-100(-)、Myoglobin(-)、CD34(-)[7,15]。细针穿刺细胞学涂片显示细胞丰富,呈单个弥散平铺分布,部分紧密聚集成团,镜下主要细胞有纤维母细胞和神经节样细胞,增生的纤维母细胞比正常纤维母细胞大,大多成群分布,常与黏液样背景同时出现,细胞大小不一,形态差异较大,多为梭形,核长且尖,核仁1~2个,染色质细,呈颗粒状,分布均匀,胞质丰富,略嗜碱性,边界清楚;神经节样细胞呈圆形或多角形,核明显增大,核仁1~2个,易见核分裂,染色质细,胞质丰富,呈嗜碱性,不整齐,背景可见粘液样,呈粉红色,部分区域可见少量中性粒细胞和淋巴细胞[16],但细针穿刺细胞学属抽样检查,有一定局限性,组织病理学才是最可靠的诊断依据。
2.6 诊断及鉴别诊断
组织病理学检查是PM的金标准,细胞学检查是诊断PM的重要依据。临床表现及体征酷似软组织肿瘤,故又称假肉瘤性病变,需与横纹肌肉瘤等软组织肉瘤相鉴别,后者系恶性肿瘤,易发生转移;超声检查与横纹肌肉瘤和纤维肉瘤具有高度相似的表现,容易误诊;组织病理学检查镜下易与结节性筋膜炎相混淆,增生性筋膜炎主要累及组织层肌束和肌纤维,应慎重仔细区分;当合并有钙化或骨化时,影像学检查与骨化性肌炎表现相似,难以区别,骨化性肌炎多有外伤,起病缓慢,病程长,无正常肌纤维成分[17]。
2.7 误诊原因
(1)PM是一种罕见病,临床极少见,医生对其认识不够,特别是在基层医院,基本没见过,即使在上级医院也难见,经验不足是误诊的重要原因。(2)病情变化快,呈浸润性生长,边界不清,本身与恶性肿瘤表现相似,导致误诊。(3)CT对软组织分辨率不如MRI,且基层医院影像医生对此缺乏经验,容易误读CT。(4)病灶呈浸润性生长,无包膜及特殊的组织学形态,大量细胞增生及细胞核分裂容易使病理医生误诊为软组织肉瘤[18]。
2.8 治疗及预后
由于PM具有自限性,一般无需积极治疗,随访即可,伴有明显症状者可服用非甾体类解热镇痛药,文献报告小剂量类固醇激素也有一定疗效,可缓解症状,虽为肌炎,但并非真正炎症,抗菌素治疗无效,对长时间不能自愈且伴功能受限者或影响正常生活者可考虑手术切除,完整切除既可治愈,术后不复发,未见恶变或转移报道,术前明确诊断十分重要,可避免因误诊导致不必要的手术。
PM是一种良性肌组织炎症性疾病,临床罕见,容易误诊,发病机制不明确,实验室检查无特异性,影像检查有典型性表现,组织病理学是诊断金标准,细针穿刺细胞学检查是重要诊断依据,但有一定局限性。误诊原因多方面,需与软组织肉瘤、结节性筋膜炎、骨化性肌炎相鉴别,其具自限性,无需过于积极治疗,预后良好。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
世界最新医学信息文摘(2021年12期)2021-06-09
世界最新医学信息文摘(2021年12期)2021-06-09
国际放射医学核医学杂志(2021年10期)2021-02-28
浙江实用医学(2019年2期)2019-06-04
中国临床医学影像杂志(2019年1期)2019-04-25
中国生殖健康(2019年9期)2019-01-07
特别健康·下半月(2017年10期)2017-10-26
