
以肺泡蛋白沉积症为首发临床表现的X-连锁高IgM综合征1例报告及文献复习
2024-01-18 01:40严士为伍秋频覃敏谭杰黄惠萍
右江医学 2023年12期
严士为,伍秋频,覃敏▲,谭杰,黄惠萍
(广西壮族自治区妇幼保健院 a.广西儿科疾病临床医学研究中心,b.儿童呼吸内科,广西南宁 530002)
由CD40配体基因(CD40 ligand gene,CD40LG)突变引起的高IgM综合征(hyper-IgM syndrome,HIgM)是一种罕见的原发性免疫缺陷病,其遗传方式为X连锁隐性遗传,CD40LG位于Xq26.3-27,其编码的CD40配体是在活化的CD4+T淋巴细胞上表达的膜结合蛋白[1]。患者以男性多见,多为 X 染色体连锁遗传,个别为常染色体隐性或显性遗传,后天获得性 HIgM 也有报道[2]。现报道1例以肺泡蛋白沉积症为首发临床表现的X-HIgM综合征,并对其临床特征及分子生物学特征进行分析,旨在提高对本病的认识,减少误诊和漏诊,提高患儿生存质量,延长寿命,对于有高危家族史的孕母,应行羊水穿刺产前基因筛查,早期干预,达到优生优育的目的。
1 病例资料
患儿男性,5个月,于2022年5月16日18时48分以“咳嗽半个月,面色差6天,气促4天”入住广西壮族自治区妇幼保健院儿童重症监护室。患儿于入院前半个月出现阵发性咳嗽,在外院予雾化治疗,咳嗽症状未见好转,6天前因面色差,肤色欠红润,继续于外院予雾化等治疗,入院前4天出现气促、呼吸费力、面色发绀、精神差、活动减少,于外院住院治疗,外院胸片提示两侧肺炎并多发实变,胸部CT提示支气管肺炎(以间质病变为主),期间予低流量吸氧、无创呼吸机辅助通气,“头孢曲松、头孢哌酮钠舒巴坦钠(舒普深)抗感染,甲强龙抗炎,人免疫球蛋白免疫支持”等治疗,患儿气促无缓解,指脉氧波动于88%~89%,血气分析:PO247 mmHg。为进一步诊治,我院出诊予气管插管呼吸机辅助通气下接回治疗。既往史:患儿既往无特殊,无传染病及密切接触史。
个人史:患儿系第3胎,第3产,胎龄37周,出生体重3100 g,系单胎。分娩方式:剖宫产(胎位不正)。生后母乳喂养,2个月停母乳,5个月添加辅食。生长发育与同龄儿相符。家族史:患儿父亲、母亲身体健康,大哥(11岁),体健,上小学4年级,成绩中等。二哥出生后3个月因“咳嗽、气促”于2016年12月22日至2017年1月22日在外院住院,诊断“重症肺炎、呼吸窘迫”等予呼吸机辅助通气、抗感染等治疗病情好转后出院(具体不详,未见出院记录);8个月大时因“气促、发绀”于2017年5月23日至2017年6月12日再次外院住院,诊断“重症肺炎、呼吸衰竭”,肺CT提示:两肺弥漫间质性病变,免疫球蛋白G 0.71 g/L,免疫球蛋白A 12 g/L,免疫球蛋白M 0.34 g/L,同时存在接种卡介苗同侧腋下淋巴结肿大。全外显子基因:当时未报致病相关基因(经再次验证患儿全外显基因存在CD40LG半合缺失)。予呼吸机辅助通气及抗感染等治疗,最终抢救无效死亡。其家族无血液病史和肿瘤病史。
体格检查:呼吸机辅助通气下,体温36.5 ℃,脉搏120 次/分,呼吸35 次/分,血压80/56 mmHg,体重6.5 kg,SPO296%,CRT 2秒。发育正常,营养良好,烦躁,精神状态差,被动体位。左上臂卡疤(+)。左腋下可触及一大小约2 cm×2 cm大小淋巴结,质地中等,边缘光滑,活动度可,余浅表淋巴结未触及肿大,呼吸平稳,无吸凹征,双侧呼吸运动对称;双肺呼吸音粗,未闻及啰音;心前区无隆起,心尖冲动正常,心率120 次/分,律齐,心音有力,未闻及病理性杂音,腹部平坦,无胃肠型、脐疝;腹软,无压痛,无腹部肿块,无肌紧张,肝脏右肋下3 cm可触及,质软,边缘锐利,脾脏未触及,叩诊呈鼓音,移动性浊音阴性;肠鸣音4 次/分;外生殖器正常,无肛裂,会阴、肛周有潮红;脊柱正常,四肢正常,关节无红肿,四肢活动自如,四肢暖,双下肢浮肿(-);肌张力正常,生理反射正常;病理反射引出,脑膜刺激征阴性。
辅助检查:血常规示白细胞(6.6~21.9)×109/L,血红蛋白(97~121)g/L,血小板(195~495)×109/L,淋巴细胞百分率(49%~93.1%),中性粒细胞百分率(2.6%~39.1%),中性粒细胞绝对值(0.17~7.61)×109/L,超敏C反应蛋白(0.56~16.02)mg/L。总T淋巴细胞(CD3+)40.53%(65%~75%),辅助/诱导T淋巴细胞(CD3+CD4+)19.27%(34%~52%),抑制/细胞毒素T淋巴细胞(CD3+CD8+)18.87%(21%~39%),辅助/抑制T淋巴细胞比值(CD4+/CD8+)1.02(0.53~2.31),总B淋巴细胞(CD3+CD19+)54.87%(9.02%~14.10%),NK淋巴细胞(CD56+CD16+)4.40%(10.04%~19.78%)。降钙素原:(0.23~0.49)ng/mL。2022年5月19日血气:酸碱度7.36,二氧化碳分压51.6 mmHg,氧分压74.8 mmHg。丙氨酸氨基转移酶(20~378)u/L,天门冬氨酸氨基转移酶(32~1507)u/L。骨髓细胞学分析:骨髓增生活跃,粒系增生减低,红、巨两系增生活跃。彩超:肝、胆、脾、胰回声未见明显异常。血串联质谱:所测氨基酸、酰基肉碱未见明显异常。尿气象质谱:未见异常。抗核抗体、抗双链 DNA、抗核糖核蛋白:阴性。真菌(1-3)-β-D葡聚糖(G试验)572.891 pg/mL(<70 pg/mL);血半乳甘聚糖试验(GM试验)0.182 pg/mL(≤0.8 pg/mL)。抗MDA5抗体:阴性。KL-6>10 000 U/mL(≤500 U/mL)。胸部CT:①两肺弥漫间质性病变,考虑肺部感染;②未除外合并肺泡蛋白沉着症;③左腋下异常密度影(大小约20 mm×21 mm×24 mm),考虑淋巴结炎并坏死可能性大(图1)。2022年5月16日(金域检验)免疫球蛋白G 0.45 g/L,免疫球蛋白A<0.2 g/L,免疫球蛋白M 1.67 g/L。总IgE<0.28 IU/mL。肺泡灌洗液PAS染色:阳性。肺泡灌洗液可见浑浊奶白色改变(图2)。肺泡灌洗液mNGS(金域检验):耶氏肺孢子菌(序列数303484)、人类疱疹病毒5型(序列数167)。左侧腋下淋巴结脓液:找到结核分枝杆菌。患儿全外显子基因检测:CD40LG基因第1-4号外显子半合缺失(chrX:135730308~135736764),OMIM:300386。
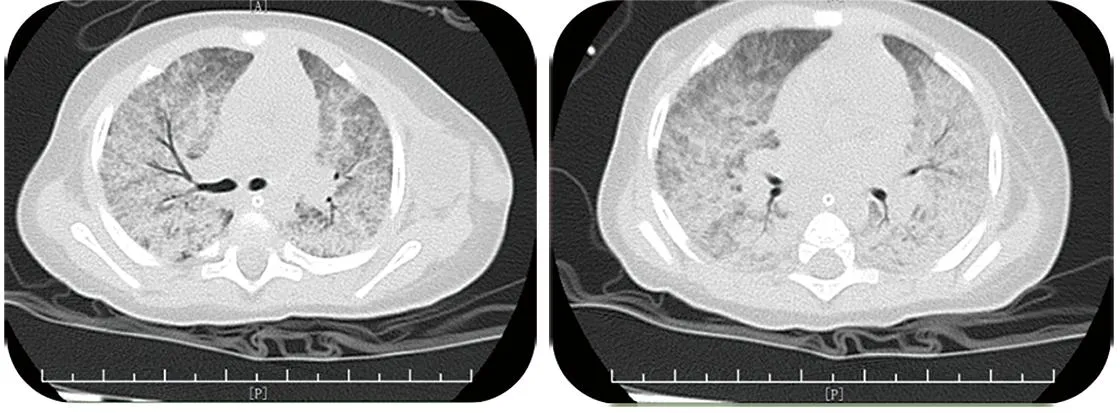
图1 患儿治疗前胸部CT影像
诊治经过:入院后予支持治疗,呼吸机辅助通气(5月16日至5月30日),CPAP辅助通气(5月30日~5月31日)、鼻导管吸氧(6月1日至出院);6月6日予转儿童呼吸内科继续治疗。肺泡灌洗液检出耶氏肺孢子菌感染后,先后予哌拉西林钠他唑巴坦、头孢哌酮舒巴坦、伏立康唑、卡泊芬净、复方磺胺甲噁唑片等治疗。住院期间予反复支气管镜分段灌洗治疗,静脉注入免疫球蛋白替代治疗。经上述治疗,患儿无气促、发绀、呼吸困难,肺泡灌洗液较前清澈(图3),肺部CT仍可见两肺弥漫性间质改变,充气较前好转(图4)。
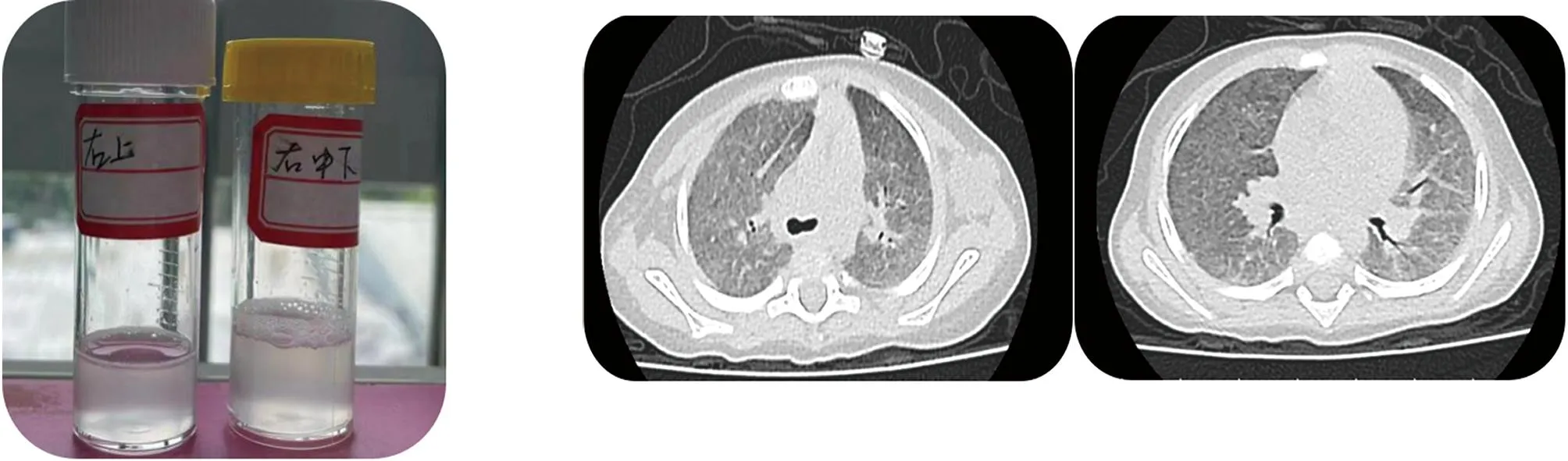
图3 患儿治疗后肺泡灌洗液 图4 患儿治疗后胸部CT影像
治疗结果、随访及转归:患儿间断低流量吸氧,无气促、发绀,出院后在专科医院行抗结核治疗。出院1个月门诊随访情况:患儿无气促、发绀,无呼吸困难,无需吸氧,左腋下淋巴结明显减小,复查肺部CT:两肺无明显弥漫性间质改变(图5)。每月来院输静注人免疫球蛋白替代治疗,因病程随访时间尚短,异基因骨髓移植尚未配型。
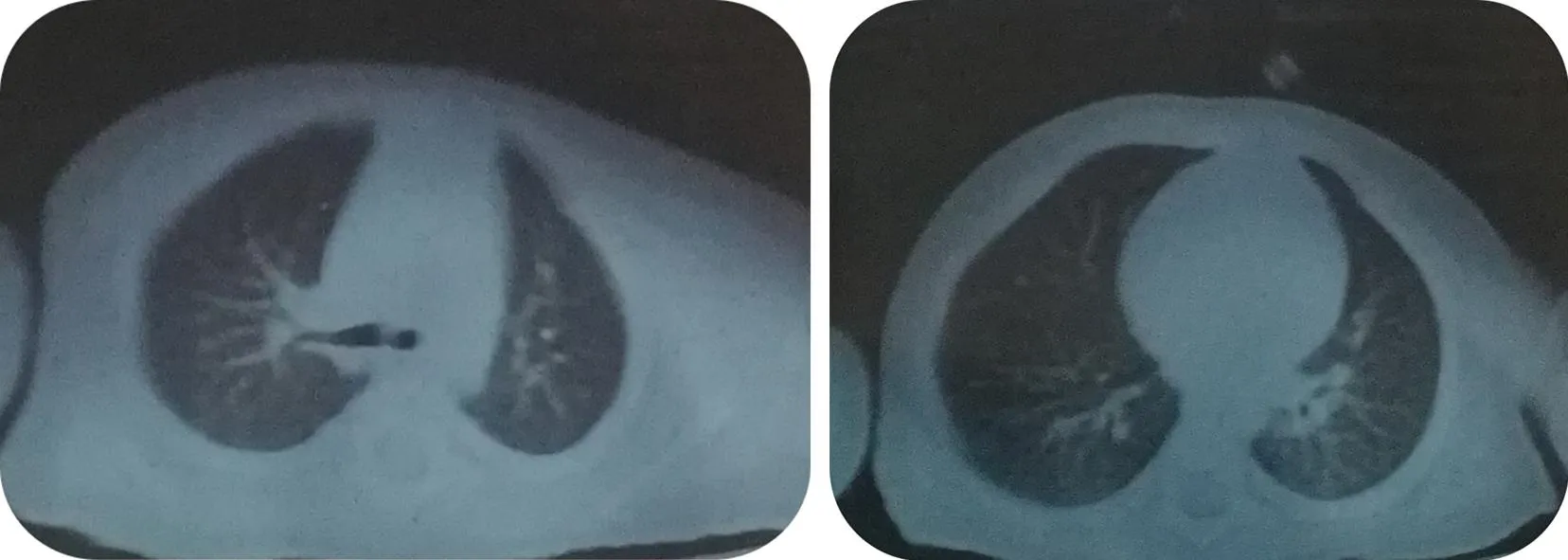
图5 出院随访胸部CT影像
2 文献复习
检索 PubMed、中国知网、万方和维普数据库,检索时间均从建库至2022年7月13日。中文关键检索词“X-连锁高IgM 综合征”,共检索出201篇,英文关键检索词“X-linked hyper IgM syndrome”,共检索出881篇。X-HIgM综合征常见的临床症状通常为:反复呼吸道感染(病毒感染性间质性肺炎、真菌性肺炎、肺泡蛋白沉积症)、噬血细胞综合征、肝功能损害、中耳炎、乳糜腹泻、胆囊炎、马尔尼菲青霉菌感染、隐球菌病、脑白质病变等。其中X-HIgM综合征合并耶氏肺孢子菌感染的文章一共15例;X-HIgM综合征以肺泡蛋白沉积症为临床表现的文章一共2例[3-4](表1)。

表1 X-连锁高IgM综合征以肺泡蛋白沉积症为临床表现病例汇总
3 讨 论
HIgM综合征是一种罕见的原发性免疫缺陷病,其特征主要是患儿血清免疫球蛋白IgG、IgA和IgE水平降低或缺乏,而血清免疫球蛋白IgM水平正常或相对升高,通常合并外周血中性粒细胞减少。临床症状复杂多样,多为反复呼吸道感染、中耳炎、胆道感染、肠道感染等,常合并条件致病菌感染,如耶氏肺孢子菌、结核分枝杆菌、非结核分枝杆菌、巨细胞病毒、弓形虫等。临床上将HIgM综合征分为5类:CD40L缺陷、CD40缺陷、活化诱导的胞苷脱氨酶(AID)缺陷、尿嘧啶DNA转葡糖基酶(UNG)缺陷以及其他,仅根据表型难以完全明确致病分子[5]。本患儿全外显基因提示CD40LG基因变异,CD40LG是肿瘤坏死因子家族的成员,主要在活化的 CD4+T细胞上表达,在B淋巴细胞增殖和生发中心形成。CD40LG突变导致缺乏记忆性B淋巴细胞和体细胞突变诱导不足,从而出现T淋巴细胞依赖性抗体反应严重不足,导致免疫缺陷,增加了条件致病感染机会[6-8]。本例患儿肺部感染不易控制,外周血淋巴细胞计数正常,中性粒细胞缺乏,体液免疫检测示免疫球蛋白G 0.45 g/L, 免疫球蛋白A<0.2 g/L,均明显低于正常范围下限,免疫球蛋白M 1.67 g/L处于正常范围,结合阳性家族史,患儿第一个哥哥正常,第二个哥哥在婴儿期发病,临床症状及肺CT跟本例患儿大致相仿,其全外显子基因与本患儿有同类变异,符合X-HIgM综合征免疫学特征,同时本患儿接种卡介苗同侧腋下淋巴结肿大、液化,淋巴结液化处脓液检出结核分枝杆菌,肺泡灌洗液中mNGS检出耶氏肺孢子,同时出现中性粒细胞缺乏,证实免疫缺陷患儿易引起条件致病感染,通常为混合感染。
肺泡蛋白沉积症(PAP)是一种由于肺泡表面活性物质代谢异常引起肺泡腔内充满大量脂质蛋白物质,造成通气功能、换气功能障碍以及肺泡巨噬细胞代谢异常,在临床上以逐渐加重的呼吸困难、气促、低氧血症、发绀、杵状指/趾为特征[9]。导致PAP的因素包括粒细胞巨噬细胞集落刺激因子信号传导受损、肺表面活性剂功能障碍综合征、血液系统疾病和其他恶性肿瘤、免疫缺陷、自身免疫性疾病、感染、药物、粉尘暴露等。根据其发病机制,PAP通常分为先天性、继发性和特发性三种类型。先天性肺泡蛋白沉积症通常是由于SP-B、SP-C、SP-D基因变异或编码GM-CSF特异性受体Bc链的基因缺陷、编码d链的基因异常(CSF2RA,CDIl6)引起肺泡蛋白沉积,从而导致新生儿肺表面活性物质代谢异常。继发性PAP 通常由于机体暴露在能够使肺泡巨噬细胞数目减少或功能受损的条件下,引起表面活性物质清除功能异常,常由免疫缺陷综合征、赖氨酸尿性蛋白耐受不良、急性硅肺病、其他吸入综合征、恶性肿瘤、感染等引起。特发性PAP是由于抗GM-CSF自身抗体竞争性地抑制内源性GM-CSF与其受体Bc链结合,阻断GM-CSF的信号转导,导致活性GM-CSF减低,引起肺泡巨噬细胞的吞噬功能、趋向能力、微生物杀灭能力下降,常在自身免疫性疾病中引起[10]。本患儿临床上有进行性气促、发绀,肺CT可见弥漫性肺间质改变,予支气管镜分段灌洗,肺泡灌洗液可见牛奶样混悬液。病理:过碘酸雪夫(PAS)染色阳性,肺泡蛋白沉积症诊断明确。本患儿肺泡灌洗液mNGS检出耶氏肺孢子菌,全外显子基因未检测到SP-B、SP-C、SP-D基因变异情况,经卡泊芬净、复方磺胺甲噁唑及多次支气管镜分段灌洗治疗,患儿气促、发绀缓解,肺部CT提示间质性改变减轻,本患儿全外显子基因提示由CD40LG变异导致X-HIgM综合征,显著降低血清中IgG、IgA,从而导致耶氏肺孢子菌感染、卡介苗相关性淋巴结炎,后继发肺泡蛋白沉积症。耶氏肺孢子菌感染引起的肺泡蛋白沉积症目前已有相关文献报道[3-4]。
先天性PAP预后不良,婴儿期常合并感染,全肺灌洗难度及风险较大,而继发性PAP预后相对较好,本患儿予静注人免疫球蛋白替代治疗,积极给予卡泊芬净、复方磺胺甲噁唑治疗耶氏肺孢子菌感染,同时予肺部分段灌洗治疗,肺部CT提示间质性肺炎可见改善(图4),无气促、发绀、呼吸困难。因此我们在临床中遇到婴儿期肺弥漫性间质性改变时,特别是同时存在接种卡介苗同侧淋巴结肿大,应尽早检测全外显子基因,除外免疫缺陷[11]。对于X-HIgM综合征的治疗,通常为每3~6周予免疫球蛋白替代治疗,根治的治疗方法主要是异基因造血干细胞移植[12-15]。X-HIgM综合征的诊断标准仍然依靠基因检测,我们可以通过临床表现以及高危家族病史,进行全外显子基因检测,从而达到早期诊断、早期替代治疗的目的,减少并发症发生,降低病死率。根据本患儿存在高危家族病史,我们可以行产前基因筛查,早期识别该类疾病,进行早期干预,从而达到优生优育的目的,最终提高我国未来人口素质。
猜你喜欢
化工管理(2022年14期)2022-12-02
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
中国生殖健康(2018年4期)2018-11-06
海洋世界(2016年3期)2016-03-24
护士进修杂志(2016年11期)2016-03-07
西南医科大学学报(2016年4期)2016-01-03
湖北农业科学(2014年11期)2014-09-10
实用中医药杂志(2014年3期)2014-03-01
