
多种形态表现的汗孔角化症一家系4例报道
2022-07-01 09:48江阳刘娟娟吕静龚娟
中国中西医结合皮肤性病学杂志 2022年3期
江阳,刘娟娟,吕静,龚娟
(重庆市中医院,重庆 400011)
汗孔角化症(Porokeratosis,PK)是一种慢性进行性、角化性皮肤遗传病,临床以边缘堤形疣状隆起、中央轻度萎缩为特点,临床上很少见。现将笔者收集的一家系4例多种形态表现的PK患者资料报道如下。
1 临床资料
1.1 病例1 先证者(Ⅰ2),患者女,74岁,以“四肢,臀部多发角化性丘疹、环状斑块伴痒50余年”为主诉。50余年前无明显诱因于四肢伸侧出现散在角化型丘疹,逐渐缓慢增大增多,向周围蔓延成不规则环状斑块,周围堤状隆起,并累及臀部,伴剧烈瘙痒。曾在外院诊断为“神经性皮炎、寻常疣”,予以抗过敏、冷冻等治疗,疗效欠佳。随年龄增长四肢皮疹反复加重,丘疹、斑块生长迅速,出现结节样损害,偶有糜烂、渗出,伴疼痛不适。双侧足底、足缘、足趾缝间出现类似损害,并融合成疣状斑块,表面粗糙皲裂。为进一步诊治于2019年2月3日来我院皮肤科就诊。既往史:高血压、2型糖尿病病史30年,自服药物控制可。皮肤科情况:四肢伸侧、臀部见较多黄豆至鸽蛋大小淡褐色斑块,中央轻度萎缩,边缘角化过度呈堤状隆起,表面少许鳞屑见图1A,双小腿胫前可见边界清楚红色糜烂面,表面少许血性渗出,中央散在花生米大小疣状结节,掌跖部位皮肤角化、皲裂、脱屑,可见疣状斑块,周围亦有环状隆起见图1B,口腔黏膜无损害。
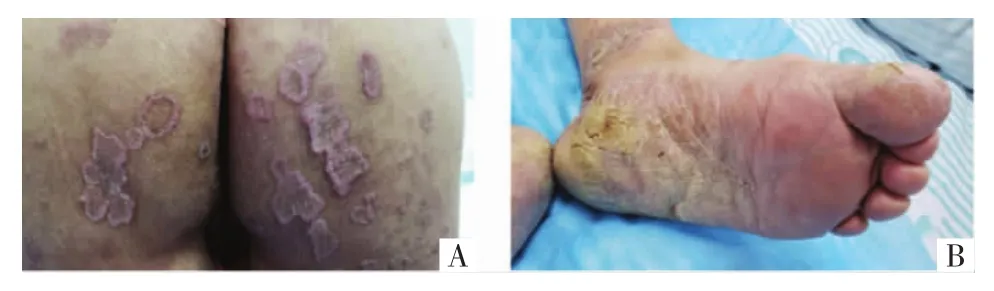
图1 先证者(Ⅰ2)皮损情况
实验室检查:血、尿常规、肝肾功、肿瘤相关因子、肝炎标志物无明显异常。臀部皮损边缘鳞屑真菌镜检为阳性,见无色分支分隔菌丝,菌种鉴定为红色毛癣菌。组织病理检查:角化过度伴角化不全,可见柱状角化不全,其下方见角化不良细胞,表皮增生肥厚,真皮浅层显著炎性细胞浸润,符合PK,见图2。
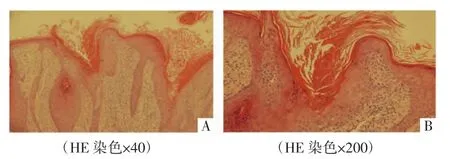
图2 先证者(Ⅰ2)皮损组织病理图
诊断:①PK。②股癣。治疗:口服盐酸奥罗他定胶囊 5 mg/次,2 次/d,雷公藤多苷片 20 mg/次,3 次/d;臀部皮损外用盐酸布替萘芬乳膏、维A酸乳膏,四肢皮损外用糠酸莫米松乳膏、苯甲酸乳膏,胫前糜烂面给予3%硼酸液湿敷。治疗后真菌镜检为阴性,斑块变平变软,颜色变淡,结节变小,糜烂面愈合,瘙痒减轻。
1.2 病例2 患者男(Ⅱ2),44岁,病例1之子,以“臀部、阴囊多发角化性丘疹、环状斑块20余年”为主诉。患者20+年前无明显诱因于双侧臀部出现角化性丘疹,不伴痛痒,缓慢向周围扩大,逐渐累及阴囊部位,发展为中央轻度萎缩,周围堤状隆起的角化过度性环状斑块,患者从未诊治。2019年3月14日因陪母亲至医院看病,追问家族史时发现患者皮疹,建议完善相关检查。皮肤科查体:阴囊部、臀部见大小不等、质地较韧的结节或疣状增生性斑块,表面角化粗糙,周围堤状隆起,边缘散在丘疹及脱屑,见图3。
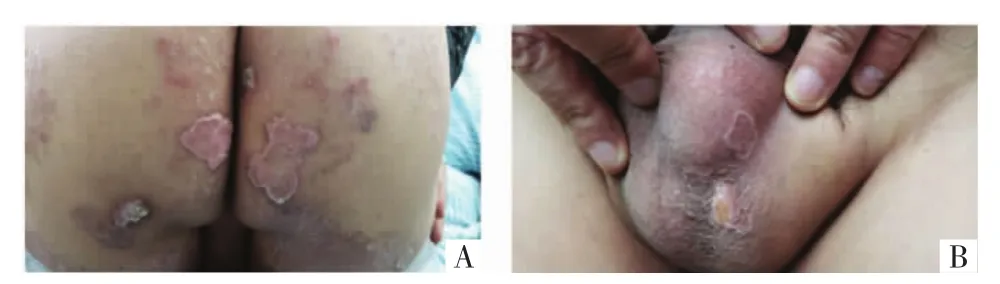
图3 病例2患者皮损照片
实验室检查:臀部皮疹真菌镜检为阳性,见无色分支分隔菌丝,菌种鉴定为红色毛癣菌。组织病理检查:角化过度伴角化不全,可见角化不良柱,其下方颗粒层消失,并可见角化不良细胞,真皮浅层周围见淋巴细胞、组织样细胞为主的炎性细胞浸润,符合PK。
诊断:①PK;②股癣。治疗方案同病例1,但患者仅同意外用药物。随访1个月皮疹稍好转,真菌镜检为阴性。
1.3 病例3 患儿女(Ⅳ1),2岁,病例1曾孙女,出生后家长即发现患儿右侧颈部、骶尾部及右下肢淡红色丘疹、斑片,随身体发育而呈比例逐渐增大。于2019年3月14日来我院就诊。查体:右侧颈部、骶尾部及右下肢见线状分布的丘疹、斑片,中央稍萎缩,边缘呈线状微隆起,部分融合成片,见图4。辅查:臀部真菌镜检阴性。因家属原因该患儿未行组织病理检查。但根据典型的临床表现和明确的家族史,诊断为PK。仅给予维生素E乳外用保湿润肤治疗,嘱其密切观察随访。病例3的母亲(Ⅲ2)大腿亦有类似皮疹,自幼发病,逐渐发展,但未提供图片。
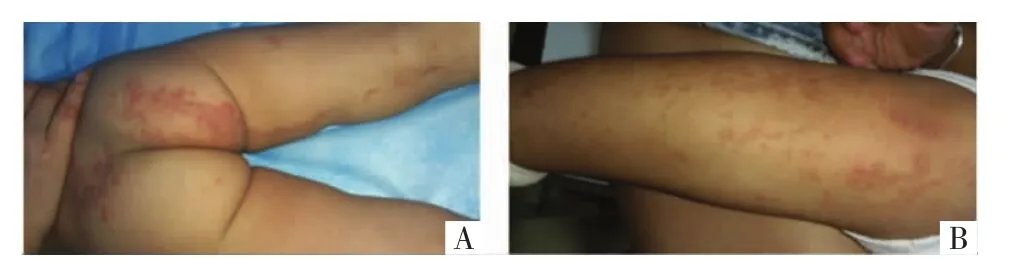
图4 病例3患者皮损照片
1.4 家系调查 该家系4代,有4例发病,其中男1例,女3例,见图5。先证者及Ⅱ2发病年龄在20岁以后,Ⅲ2、Ⅳ1均为自幼发病,目前仍有发展的趋势。皮疹主要累及四肢、臀部、掌跖等部位,随年龄的增长、曝光时间的延长及局部受压摩擦等刺激,患者皮疹呈进行性发展和加重,初起为边缘略微线状隆起的环状丘疹、斑片,逐渐发展为疣状增生的斑块、结节,中央萎缩,边缘堤状隆起,伴或不伴瘙痒,高龄患者应警惕皮疹癌变的可能。该家系有以下遗传特点:4代中每代均有患病,男女均可发病,患者必有双亲之一患病,连续传代,无病的子女与正常人结婚,其后代一般不再有此病。这符合常染色体显性遗传病的特点。

图5 患者家系图
2 讨论
PK是一种较少见的遗传性角化性皮肤病,多呈常染色体显性遗传,有家族聚集现象,可在一个家庭几代成员中发病,但也有无遗传证据的散发病例[1]。同一家系中发病的患者可存在发病年龄和皮损轻重程度的不同[2]。该病为表皮细胞在某些刺激下的不正常克隆性增生,有癌前倾向,免疫组化方法已经证明了皮损中p53蛋白呈过度表达[3]。依据目前所发现的突变基因或位点可将PK分为9个基因型,每一型可包含不同的临床表型[4],各型PK的临床表现差异较大,这可能和不同的基因突变有关[5-6],但其共同特征是角化不全细胞柱(角样板层),提示其发病模式非常相似。部分患者可伴有鳞状细胞癌、银屑病、角化性湿疹、皮肤真菌感染,提示遗传、环境及免疫因素在PK的发病中起着重要的作用。
多数学者认为损害是由细胞遗传学异常的细胞株构成,当正常的免疫过程被破坏后此细胞株得以增殖,即导致发病[7]。该病的诊断要点为:①基本皮损为角化性环形或不规则形棕褐色丘疹、斑块,中央略萎缩凹陷,周围呈堤状隆起;②好发于四肢、面部等,一般有家族史,近年来会阴、臀部等处发病者亦有增多;③特征性组织病理可见角化不全柱[8]。笔者报道的4例有家族史的PK患者,年龄依次为婴幼儿、青年、中年、老年,可以观察到该家族中不同年龄阶段PK的基本临床特点,婴儿时期的皮疹以粟粒至黄豆大小的角化性丘疹为主,周围呈线状微隆起,皮损沿肢体单侧线状分布;中年后的皮疹以角化性丘疹、斑块为主,周围见典型的堤状角质型隆起,累及臀部、阴囊等处,无明显自觉症状;老年后的皮疹主要累及四肢、臀部,除角化性丘疹、斑块、堤状隆起以外,还可出现结节、破溃,伴剧烈瘙痒,需警惕发生皮损癌变的可能。从这4例病例可以看出,PK是一种慢性的皮肤病,临床发展缓慢,可逐渐出现增生、结节、破溃,甚至癌变。发生在肛周、臀部、生殖器周围的角化性斑块,要考虑到疣状PK的可能(如Ⅰ2、Ⅱ2的皮疹)。国外有学者认为巨大型、病程长、线状PK是恶变的高风险类型(和遗传学上杂合性缺失有关)[9],故Ⅳ1患儿需密切随访,必要时说服家长同意组织病理学检查。马蕾等[10]曾报道1例患者,随年龄增长相继出现不同的PK临床表现,包括多发性斑片、角化性结节、疣状增殖性斑块等,这也说明了不同年龄阶段PK的临床表现各有特点。本病临床上需与疣状皮肤结核、疣状扁平苔藓、疣状痣、着色真菌病、银屑病、红斑狼疮、慢性湿疹等疾病鉴别[11],呈线状排列时还需与线状苔藓、线状扁平苔藓相鉴别。前者的板层状角化不全柱是主要鉴别要点。
PK的组织病理特征为:在充有角蛋白的凹陷中央有一角化不全栓(圆锥形板层),其下方的表皮角质形成细胞排列不规则。其角化不全柱与皮损边缘线状环堤状嵴样隆起相对应[12],受压或者摩擦部位的皮疹由于局部的物理刺激及搔抓等因素,常可出现斑块型疣状损害及结节、破溃等,如发生恶变则有相对应的组织病理变化。本文中第1例和第2例患者臀部环状皮疹真菌镜检均为阳性,菌种鉴定为红色毛癣菌,该菌是最常见的嗜角蛋白皮肤癣菌之一,感染后可使皮损处角质堆积,而大量的角质堆积又使得红色毛癣菌进一步滋生。由此推测红色毛癣菌可能是表皮细胞不正常克隆增生的激发因素之一,国内曾有类似报道,但红色毛癣菌是否和PK的发病有直接关系目前尚不明确,可能主要作为一种刺激因素存在[13]。
本病治疗无特异性,即使消退仍可能复发,基本原则是对症处理。液氮冷冻、二氧化碳(CO2)激光、光动力、手术切除等方法可以选用[14]。文献报道PK的恶变率为7.5%~11.0%,可发展为鲍温病、基底细胞癌、鳞状细胞癌[15],因此早期诊断和早期治疗非常重要。皮损面积广泛时可选择口服维甲酸类药物治疗。寻找致病基因,对该疾病进行产前诊断、基因治疗,是今后主要的研究方向。
猜你喜欢
纺织高校基础科学学报(2022年2期)2022-07-29
医学前沿(2021年6期)2021-09-10
数学学习与研究(2021年18期)2021-08-06
家庭科学·新健康(2020年6期)2020-07-06
中国美容医学(2019年2期)2019-03-13
东方教育(2017年14期)2017-09-25
课程教育研究·学法教法研究(2016年1期)2016-03-17
右江医学(2014年1期)2014-03-22
疯狂英语·口语版(2013年8期)2013-08-19
为了孩子(孕0~3岁)(2001年24期)2001-01-23
