
先天性丛状血管瘤1例
2024-01-08 01:10何萍秀程丽芳吴忆夏碧霞
皮肤性病诊疗学杂志 2023年6期
何萍秀, 程丽芳, 吴忆, 夏碧霞
江西省皮肤病专科医院 江西省皮肤病临床医学研究中心,江西 南昌 330001
1 临床资料
患儿男,5岁,因右肩胛部暗红色斑块5年就诊。患者出生时发现右肩胛部有一约1元硬币大的暗红色斑,表面光滑,随年龄逐渐增大、增厚,稍隆起于皮肤。1岁时红斑中央开始逐渐向下凹陷,无破溃,无自觉症状,未曾治疗。患儿系第一胎第一产,足月顺产,否认手术外伤史及家族遗传性疾病史。体格检查:患儿发育正常,各系统检查未见异常。皮肤科检查:右肩胛部见一约5.0 cm×4.0 cm的暗红色浸润性斑块,表面光滑,中央略凹陷,边缘呈堤状隆起(图1)。实验室检查:血常规及凝血四项均未见异常。皮损组织病理检查:表皮棘层不规则肥厚,皮突延长。真皮全层见散在分布多个大小不一的肿瘤细胞团块,主要由毛细血管和大小均一的梭形或类圆形内皮细胞组成,成“炮弹”样分布(图2A、2B)。免疫组织化学染色示:CD31(+)(图3),CD34(+),Ki-67(+,<1%)。诊断:先天性丛状血管瘤。行皮损扩大切除加皮瓣成形术,术后恢复良好。随访1年半,无复发。
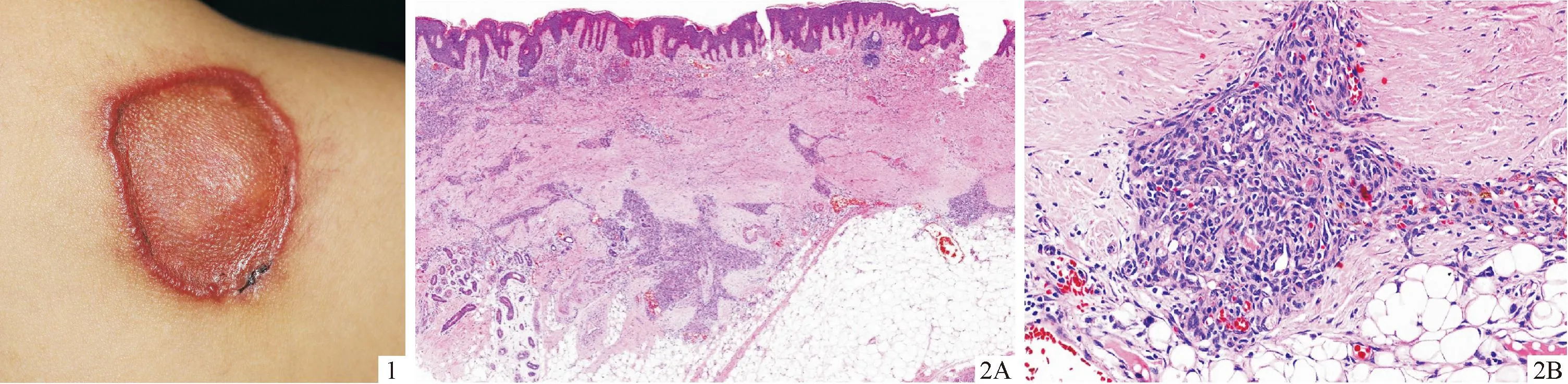
图1 患者右肩胛部暗红色浸润性斑块,边缘呈堤状隆起,中央凹陷 图2 皮损组织病理检查(HE染色) 2A:真皮内散在分布多个大小不一的肿瘤细胞团块,形态不规则(20×);2B:增生的毛细血管及内皮细胞紧密排列呈结节状(200×)

图3 免疫组织化学染色:CD31(+)(SP法,200×)
2 讨论
丛状血管瘤(tufted angioma,TA)是一种罕见的良性血管肿瘤,通常位于皮肤和皮下组织,发病机制尚不明确[1]。本病好发于婴幼儿或儿童[2],先天性发病者高达72%(39/54)[3],个别报道成人起病[4]。TA无明显性别差异,多见于躯干上部、颈部及四肢近端,临床表现为大小不等的暗红色至紫罗兰色丘疹、斑疹或斑块,单发或多发,病变通常无自觉症状,部分伴疼痛、压痛、瘙痒、多毛、多汗症等[3]。大多数患者的皮损随身体生长而缓慢增大,少数自发部分或完全消退[5]。本例患儿出生后皮损即出现且随身体生长而增大,1岁后开始斑块中央萎缩凹陷,边缘呈堤状隆起,考虑该患者皮疹中央出现部分自行消退现象。
TA临床表现无特异性,诊断依据组织病理学检查及免疫组化标记。组织病理改变为真皮和皮下组织浅层散在毛细血管组成的圆形、卵圆形或细长形团块,低倍镜下呈“炮弹样”外观,团块边缘可见新月形或半月形的扩张淋巴管[2]。免疫组化示增生的血管内皮细胞表达CD31、CD34,周围扩张的淋巴管表达D2-40[6],不表达GLUT-1。本病组织学上需与婴儿血管瘤、小叶性毛细血管瘤、卡波西样血管内皮瘤(kaposiform hemangioendothelioma,KHE)等具有团块状增生的血管瘤鉴别。婴儿血管瘤的真皮及皮下组织受累更为弥漫、融合及广泛,表达GLUT-1。小叶性毛细血管瘤好发于头颈部和四肢,皮损为有蒂或无蒂的红色丘疹或结节,易发生溃疡和出血,组织学上病变两侧表皮增生形成“领圈”样结构,真皮内常伴炎细胞浸润,二者不难鉴别。有学者报道TA和KHE可能为同种血管肿瘤的不同阶段,两者可相互转化[7],临床及病理特征相似,如出现向周围组织浸润性生长、肾小球样结构、含铁血黄素沉积和透明小球,则倾向于诊断KHE。
高达10%的TA可伴有Kasabach-Merritt现象(Kasabach-Merritt phenomenon,KMP)[8]。KMP是在脉管性疾病基础上伴发血小板减少、微血管溶血性贫血和消耗性凝血功能障碍的一类临床表现,可发生于罕见的血管肿瘤KHE和TA[6],其病程凶险,可危及生命,死亡率高达20%。因此,伴有KMP的TA需要及早治疗[9]。
本病目前尚无特效的治疗方法。由于部分病例可能自发消退,有学者认为应定期监测病变[10]。然而大部分病例无自然消退倾向,对于浅表、局限的病灶首选手术切除。对于病灶体积大、浸润广泛、界限不清及合并KMP的病例,可使用药物有效控制病灶体后,再行手术根治[6]。文献报道治疗有效的药物主要有皮质类固醇、普萘洛尔、长春新碱、雷帕霉素[9,11-12]等。本例患者不伴有KHP,皮损中央部分消退,行手术完整切除后随访1年半无复发。
猜你喜欢
世界最新医学信息文摘(2022年8期)2022-07-27
西安石油大学学报(自然科学版)(2021年2期)2021-04-23
中国肿瘤临床(2020年19期)2020-11-23
都市(2018年5期)2018-09-10
中南大学学报(自然科学版)(2016年2期)2017-01-19
作文新天地(初中版)(2016年3期)2016-03-18
作文新天地(2016年15期)2016-02-11
河北医药(2015年13期)2015-11-13
新疆钢铁(2015年3期)2015-11-08
中国现代药物应用(2015年19期)2015-01-23
